Секвенирование простых геномов
Секвенирование простых геномов
Для определения нуклеотидной последовательности (т. е. первичной структуры) конкретного района ДНК в первую очередь необходимо упростить ее, что достигается путем разрезания ее на относительно короткие фрагменты. Сделать это можно, например, с помощью специальных «скальпелей» для ДНК — ферментов рестриктаз, о которых уже шла речь выше.
При секвенировании простейших организмов, у которых геном относительно невелик, обычно используют процедуру, называемую условно «сверху вниз». Всю ДНК «разрезают» на кусочки с помощью уже упоминавшихся выше ферментов рестриктаз, затем секвенируют эти кусочки по отдельности, а после «склеивают» из них полный геном. «Склеивание» оригинала осуществляется за счет того, что нуклеотидные последовательности разных кусочков перекрываются друг с другом, т. е. одинаковы по концам. Эта методология получила название «дробовика». Суть данной процедуры отражена на рис. 16.
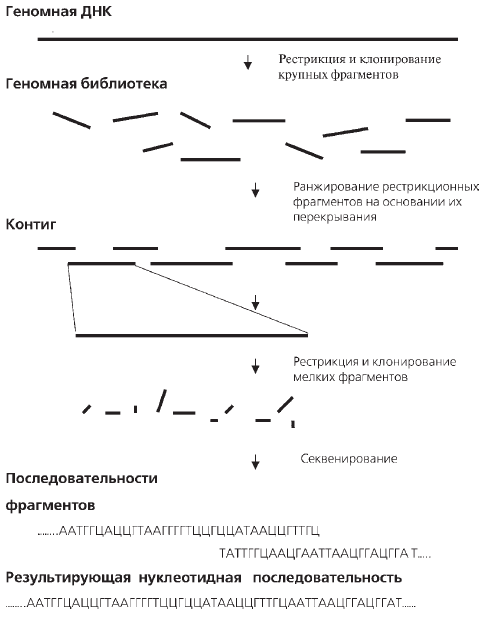
Рис. 16. Схема стратегии «дробовика», используемая для секвенирования больших молекул ДНК
Однако в случае такого очень сложного генома, как геном человека, начали с другого, а именно с определения положения известных генов и других генетических маркеров на отдельных хромосомах, то есть с генетического картирования генома. Подобную задачу генетическими экспериментами на более простых объектах пытался решить еще Т. Морган, который за свои работы получил Нобелевскую премию в 1933 году. Теперь появились более эффективные методы. Один из них, называемый методом «радиационных гибридов», заключается в следующем. Клетки человека, растущие вне организма в питательной среде, облучают рентгеном, что приводит к гибели клеток в результате разрыва хромосомной ДНК на куски, содержащие от 2,5 до 25 млн. п. н. Но прежде, чем убитые облучением клетки распадутся, их сливают с клетками хомяка, в результате чего в разные клетки хомячка попадают разные наборы фрагментов ДНК человека. Затем «гибридные» клетки размножают в культуре, при этом в них наряду с собственной ДНК реплицируются и фрагменты чужеродной ДНК. Затем определяют состав известных генов и других генетических маркеров в каждой клеточной линии и, статистически обработав полученные данные, выводят наиболее вероятное их взаимное расположение в хромосомах. В качестве генетических маркеров использовали как гены, так и фрагменты ДНК с неизвестной функцией. Для картирования хромосом важным свойством маркеров являлся их полиморфизм, т. е. существование разных форм среди индивидуумов.
Так были построены первые генетические карты генома человека, на которых первоначально были отмечены различные генетические маркеры, отстоящие друг от друга на расстоянии не более 2 миллионов нуклеотидных пар (млн. н. п.).
Затем были составлены физические карты хромосом: первоначально с разрешением 0,1 млн. н. п., а затем 0,001 млн. н. п. Для этой цели на первом этапе применяли методы окрашивания хромосом и гибридизации с хромосомами in situ. И лишь позднее использовали рестриктазы. С помощью этих удивительно точно работающих ферментов «дробили» ДНК по строго определенным участкам на миллионы перекрывающихся между собой по нуклеотидной последовательности фрагментов, «разбирали» каждый из них по отдельности, после чего из них «склеивали» оригинал. «Склеивание» проводили на основании перекрывания фрагментов по нуклеотидной последовательности. Так постепенно шли все выше и выше. Поэтому данная стратегия получила название «снизу вверх». Со всей очевидностью можно сказать, что решалась грандиозная по масштабам и сложности задача. И решить ее ученые смогли только с помощью суперкомпьютеров. В результате были созданы физические карты разных областей ДНК и целых хромосом, состоящие из последовательно перекрывающихся друг с другом фрагментов. Набор таких «родственных» фрагментов получил название контиг (рис. 16).
А далее наступила очередь секвенирования отдельных рестрикционных фрагментов. Это привело в конечном итоге к построению секвенсовых карт, на которых степень разрешения была доведена до своего максимального значения. Если 20 лет назад расшифровка нуклеотидной последовательности ДНК длиной 1000 п. н. считалась большим научным достижением (за это можно было сразу получить степень доктора наук), то уже к 1990 году секвенирование ДНК стало массовой технологией. А сейчас квалифицированный лаборант проделывает такую работу всего за несколько часов.
После появления эффективных методов секвенирования ДНК и нескольких стратегий использования этого метода стал стремительно нарастать вал расшифрованных нуклеотидных последовательностей, в первую очередь таких простых организмов, как вирусы, а также отдельных клонированных фрагментов ДНК различных видов высших организмов. Так, еще в конце 1970-х годов была расшифрована структура первого живого объекта — вируса бактерий — бактериофага, обозначаемого как ?? 174, имеющего длину 5386 п. н. Затем последовала очередь других.
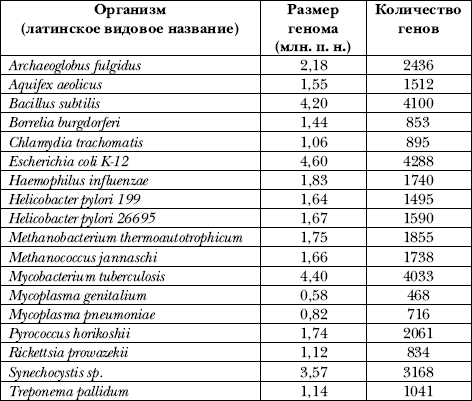
В конце 80-х гг. были начаты крупные международные проекты, целью которых было полное секвенирование геномов бактерий, грибов, дрожжей, дрозофилы, мыши, пшеницы, человека и др. Первоначально была определена первичная структура ДНК микроорганизмов с размерами генома до 20 млн. п. н., к концу 1998 г. их число составило уже 18 (см. табл. 2). Наименьший размер генома у свободно живущих организмов (например, у бактерии Mycoplasma genitalium) составляет лишь около 600 000 п. н. У этой бактерии содержится всего 500 генов, причем 150 из них (если их удалять по одиночке) никак не влияют на её жизнеспособность. Так возникло предположение, что элементарная «машина жизни» теоретически должна работать при наличии всего лишь 350 генов.
Первые объекты для секвенирования были выбраны неслучайно. Большинство из этих микроорганизмов (архебактерии, спирохеты, хламидобактерии, кишечная палочка, возбудители пневмоний, сифилиса, гемофилии, метанобразующие бактерии, микоплазмы, риккетсии, цианобактерии) способны вызывать различные патологии у человека. В настоящее время многие из этих проектов уже завершены; исследовано свыше 800 полных клеточных геномов микоплазм, архебактерий, кишечной палочки, возбудителей ряда болезней человека, а также пекарских дрожжей, маленького червя-нематоды, дрозофилы и весьма интересного в практическом плане растения арабидопсиса. Весьма вероятно, что истинное число секвенированных к настоящему времени геномов гораздо больше, потому что многие фармацевтические фирмы засекречивают свои результаты, не публикуя их в открытой печати.
Таблица 2. Некоторые микроорганизмы, геномы которых полностью секвенированы к настоящему времени
Более 800 000 книг и аудиокниг! 📚
Получи 2 месяца Литрес Подписки в подарок и наслаждайся неограниченным чтением
ПОЛУЧИТЬ ПОДАРОКДанный текст является ознакомительным фрагментом.
Читайте также
Секвенирование простых геномов
Секвенирование простых геномов Для определения нуклеотидной последовательности (т. е. первичной структуры) конкретного района ДНК в первую очередь необходимо упростить ее, что достигается путем разрезания ее на относительно короткие фрагменты. Сделать это можно,
8.4. Эволюция генов и геномов
8.4. Эволюция генов и геномов Анализ структуры и изменчивости генетического материала служит основой для различных теорий эволюции гена как элементарного носителя генетической информации. Какова была исходная организация гена? Или, другими словами, обусловлены ли
ОБЩЕЕ ВВЕДЕНИЕ ПОЧЕМУ ДЛЯ РЕШЕНИЯ СЛОЖНЫХ ВОПРОСОВ ДОСТАТОЧНО ПРОСТЫХ ИССЛЕДОВАНИЙ
ОБЩЕЕ ВВЕДЕНИЕ ПОЧЕМУ ДЛЯ РЕШЕНИЯ СЛОЖНЫХ ВОПРОСОВ ДОСТАТОЧНО ПРОСТЫХ ИССЛЕДОВАНИЙ В этой книге я предлагаю провести семь экспериментов, которые могли бы изменить наше представление об окружающем мире. Эти эксперименты способны вывести нас за пределы современного
Таблица 6. Характеристика полностью расшифрованных геномов ряда про– и эукариотических организмов (по B. Alberts et al, 2002, Molecular biology of the cell)
Таблица 6. Характеристика полностью расшифрованных геномов ряда про– и эукариотических организмов (по B. Alberts et al, 2002, Molecular biology of the
Глава 9 Метод дробовика – шотган-секвенирование
Глава 9 Метод дробовика – шотган-секвенирование Если вы не можете объяснить любому, что вы делали, значит, вы занимались полной ерундой. Эрвин Шрёдингер Итак, мы научились выявлять гены человека с невообразимой скоростью, но наши достижения только разожгли аппетит