Глава 2 От синтетической теории эволюции к эволюционной геномике: различные механизмы и пути эволюции
Глава 2
От синтетической теории эволюции к эволюционной геномике: различные механизмы и пути эволюции
Пер. А. Нестеровой
В этой главе мы продолжим обсуждение эволюционной биологии в период до появления геномики. Многие из обсуждаемых направлений развития не являлись предшественниками синтетической теории эволюции (СТЭ). На самом деле они возникали параллельно с развитием СТЭ, но были отвергнуты «каноном» вследствие «ужесточения» СТЭ. Достижения, которые обсуждаются в этой главе, относятся к интервалу между 1930 (публикация книги Рональда Фишера, которая ознаменовала вторую, зрелую стадию развития эволюционной биологии) и 1995 годами (первые сравнения полных геномов клеточных форм жизни). Моя цель здесь — вкратце обрисовать сложную сеть эволюционных идей, теорий и наблюдений, которые дополнили достаточно жесткую структуру СТЭ и стали пусковой площадкой для нового, «геномного» подхода к изучению эволюции.
Репликация цифровых носителей информации: центральный принцип биологии и необходимое и достаточное условие эволюции
Модель структуры ДНК, представленная Джеймсом Уотсоном и Фрэнсисом Криком (очевидно, основанная на рентгеновских структурах, полученных Розалинд Франклин и другими), несомненно, является одним из главных открытий не только биологии XX века, но и всей истории биологии (Watson and Crick, 1953b). Однако этот прорыв не всегда упоминается в связи с принципами биологической эволюции.
С моей точки зрения, структура ДНК и модель ее репликации, которую Уотсон и Крик описали в своей второй классической статье как непосредственное следствие структуры (Watson and Crick, 1953a), являются важнейшим фундаментальным открытием в изучении эволюции со времени публикации «Происхождения видов…». По сути, Уотсон и Крик вывели из структуры ДНК биологическое воплощение общего принципа цифрового хранения, кодирования и передачи информации. Система биологической передачи информации, которую выявили их исследования, может рассматриваться как расширение принципа машины Тьюринга, сначала через правила комплементарности нуклеотидных оснований (в процессах репликации и транскрипции), a затем в процессе трансляции, через генетический код (см. рис. 2-1). По сути, пусть и не в историческом смысле, эти открытия вытеснили концепцию Дарвина, в том смысле, что вся дарвиновская схема эволюции является прямым следствием механизма репликации ДНК. Для всех известных форм жизни биологическая передача цифровой информации влечет за собой исполнение следующих простых фундаментальных принципов[15].
Генетический материал любого организма состоит из линейной последовательности символов, четырех оснований нуклеиновых кислот, которая, прямо или косвенно, кодирует всю информацию, необходимую для построения организма[16].
Репликация генетического материала, являющегося механической основой наследственности, осуществляется на основе принципа однозначного комплементарного соответствия между A и T(U), и G и C. (Так называемые правила Чаргафа, по имени их первооткрывателя, австрийского, a затем американского химика Эрвина Чаргаффа[17].)
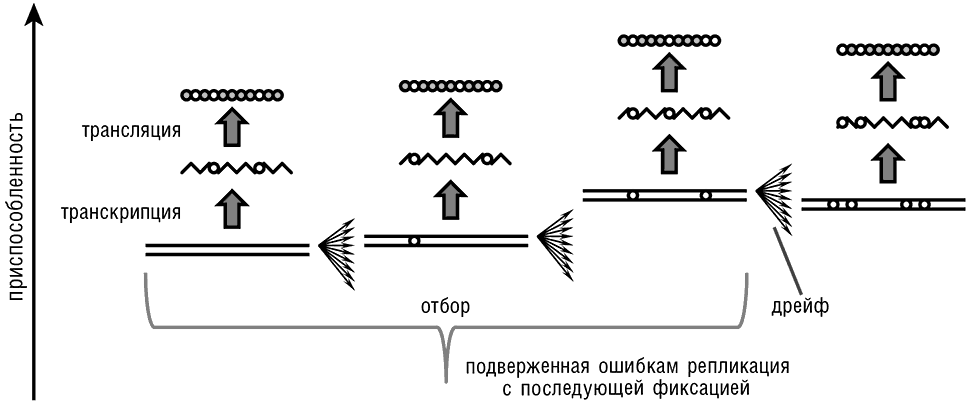
Рис. 2-1. Передача информации в биологических системах и превращение естественного отбора и генетического дрейфа в эпифеномены репликации. Белые круги на схеме обозначают изменения относительно оригинальной последовательности.
Уотсон и Крик описали эти ключевые принципы генетических систем в двух своих статьях, вышедших в 1953 году. Дальнейшие исследования добавили два очень важных аспекта:
1. Принцип комплементарности используется не только во время репликации, но и во время транскрипции ДНК во все виды РНК и во время трансляции мРНК в белок с помощью адапторных молекул тРНК.
2. Те же самые принципы цифровой репликации и декодирования применимы и для генетических систем, в которых генетический материал отличается от двойной спирали ДНК, изначально описанной Уотсоном и Криком, и состоит из РНК или односпиральной ДНК (например, у многих вирусов).
Теория информации твердо стоит на том, что передача информации абсолютно без ошибок невозможна в принципе. В реальности вероятность ошибки в любом конечном сообщении может быть сведена к минимуму, но любое снижение уровня ошибок при передаче информации возможно только за счет затраты энергии. Эта связь непосредственно следует из законов термодинамики. Центральный принцип эволюции может быть сформулирован следующим образом:
Репликация цифровых носителей информации неизбежно подвержена ошибкам, что влечет за собой эволюцию этих носителей путем естественного отбора и случайного дрейфа генов при условии, что уровень ошибок репликации ниже катастрофического порога, имеющего порядок величины от одной до десяти ошибок на геном за один цикл репликации.[18]
Назовем это обобщение принципом подверженной ошибкам репликации (ПОР)[19]. Этот принцип становится самоочевидным, как только мы осознаем существование и основной механизм репликации. Он был впервые описан математически в теории Манфреда Эйгена (Eigen, 1971), который также ввел понятие концепции порога ошибки (Biebricher and Eigen, 2005) — эта теория и ее применение будут рассматриваться далее в главе 12. ПОР основывается на следующих двух предположениях, которые могут показаться очевидными, но заслуживают тем не менее особого упоминания:
1. Ошибки репликации наследуются (проходят через циклы репликации).
2. Существует обратная связь между генотипом и фенотипом: некоторые ошибки репликации влияют на эффективность и точность репликации как отрицательно, так и положительно.
Это и отличает биологические репликаторы с их «неограниченной наследуемостью» от репликаторов с «ограниченной наследуемостью», таких как кристаллы или ряд химических циклов, которые реплицируются, но не передают накопленные дефекты последующим поколениям (Szathmary, 2000). Упрощенно говоря, разница в том, что в нуклеиновых кислотах замена одного нуклеотида на другой влияет только на передаваемую информацию, а не на физические или химические свойства носителя информации (во всяком случае, не значительно), как в случае небиологических систем.
В принципе должен существовать и нижний порог частоты ошибок репликации. Очевидно, что если математическое ожидание числа ошибок на цикл репликации стремится к нулю, то разнообразие становится недостаточным для эволюционного процесса. Однако необходимо заметить, что уровень ошибок репликации нуклеиновых кислот, определяемый эмпирически, в отсутствие сложных корректирующих механизмов (как, например, в РНК вирусов), не намного ниже верхнего, катастрофического порога. Таким образом, необходимым условием эволюции является достаточно низкий (а не достаточно высокий) уровень ошибок репликации[20]. Вопрос о том, в какой степени оптимизируется фактическая точность передачи информации в биологической системе (другими словами, эволюционирует ли эволюция), весьма сложен, интересен и широко обсуждается. Мы рассмотрим его подробнее в главе 9.
Несмотря на то что вся естественно эволюционировавшая жизнь основана на репликации нуклеиновых кислот, ПОР не зависит от физической природы репликаторов, как можно видеть на примере эволюции компьютерных вирусов и различных компьютерных моделей эволюции искусственной жизни (Lenski et al., 2003). Тем не менее вопрос о том, необходим ли цифровой код для эволюции или эволюция может происходить и в аналоговых системах, весьма интересен и до сих пор остается открытым.
В главе 1 был затронут вопрос o псевдотавтологичной природе естественного отбора. По сути, ПОР действительно в значительной мере тривиализирует естественный отбор и генетический дрейф (два фундаментальных принципа эволюции), как бы отбирая у них статус независимых феноменов и низводя их до эпифеноменов ПОР. Это ни в коей мере не умаляет достижения Дарвина, Райта и других выдающихся эволюционных биологов и не уменьшает важность концепций естественного отбора и генетического дрейфа для описания эволюционных процессов на абстрактном уровне. Тем не менее открытие репликации с контролируемым уровнем ошибок обнаруживает более фундаментальные принципы, которые лежат в основе классических положений эволюционной биологии.
Молекулярная эволюция и молекулярная филогенетика
Традиционные филогенетические исследования, наполнившие содержанием дарвиновскую концепцию древа жизни, были основаны на сравнении морфологических черт организмов, таких как структура скелета животных или строение цветков растений (Futuyma, 2005). Эволюционные биологи не осознавали, что сравнивать следует реальную молекулярную базу эволюции, которая подвержена действию естественного отбора, то есть гены, просто потому, что они практически ничего не знали о химической основе этой составляющей и о способе, которым гены кодируют фенотип организма. Более того, согласно парадигме панадаптационизма в эволюционной биологии, гены, на каком бы молекулярном механизме они ни основывались, должны существенно разниться в неродственных организмах, если учесть фенотипические различия между этими организмами, как подчеркивалось, в частности, Эрнстом Майром, одним из главных строителей СТЭ.
Идея того, что последовательность оснований ДНК может использоваться для эволюционной реконструкции, была, вероятно, впервые высказана в печати, пусть и мимоходом, еще Криком (в той же самой основополагающей статье, в которой он сформулировал адапторную гипотезу синтеза белка, — Crick, 1958). Эмиль Цукеркандль и Лайнус Полинг развили принципы и описали первое фактическое использование молекулярного эволюционного анализа несколько лет спустя. Они напрямую опровергли гипотезу Майра, показав, что последовательности аминокислот нескольких белков, которые в то время были известны для нескольких видов, такие как цитохром c и глобины, оказались чрезвычайно консервативными даже у дальнородственных животных (Zuckerkandl and Pauling, 1965). Цукеркандль и Полинг также предложили концепцию молекулярных часов: они предсказали, что скорость эволюции определенной последовательности белка будет неизменна (с учетом возможных флуктуаций) в течение длительных временных интервалов в отсутствие функциональных изменений. Здесь необходимо отметить, что то, что последовательности генов, кодирующих «один и тот же белок» (то есть белки с одинаковым действием и сходными свойствами), в различных организмах оказались очень сходными — и, более того, степень схожести этих последовательностей отрицательно коррелировала с филогенетическим расстоянием между данными организмами, — можно рассматривать как наилучшее и исчерпывающее доказательство реальности эволюции.
В течение последующих нескольких лет, в основном благодаря работам Маргарет Дэйхофф и ее коллег, консервативность кодирующих белки последовательностей была продемонстрирована на примерах самых различных форм жизни, от бактерий до млекопитающих (Dayhoff et al., 1983). Учитывая открытие консервативности белок-кодирующих последовательностей и гипотезу молекулярных часов, оказалось естественным перейти к конструированию филогенетических деревьев на основе (не)похожести этих последовательностей, что должно было показать примерное время возникновения расхождений в последовательностях генов (белков) от общего предка. И в самом деле, скоро были изобретены несколько методов измерения расстояний в молекулярной филогенетике, а также введен принцип наибольшей экономии (см. табл. 2-1). Последующее тестирование гипотезы молекулярных часов на все растущей базе последовательностей ДНК показало, что для большинства генов эти часы идут не с одинаковой скоростью; напротив, эти данные оказались значительно диспергированы, то есть отклонения в скорости эволюции значительно превышали среднее отклонение, которое могло быть предсказано распределением Пуассона (Bromham and Penny, 2003). Такая избыточная дисперсия молекулярных часов приводит к особенности молекулярного филогенеза, известной как притяжения длинных ветвей (ПДВ), существенно искажающей результаты молекулярного филогенетического анализа (см. табл. 2-1). Молекулярная филогенетика, таким образом, превратилась в сложную ветвь прикладной математики и статистики в основном для того, чтобы справляться с эффектами ПДВ и им подобными (Felsenstein, 2004). Но, несмотря на все артефакты, молекулярная филогенетика остается краеугольным камнем современной эволюционной биологии и использует в первую очередь методы наибольшего правдоподобия (см. табл. 2-1).
Таблица 2-1
Краткое описание филогенетических методов
Методы, основанные на секвенировании
Требуют многократного сравнения гомологичных нуклеотидов или белковых последовательностей.
Дистанционно-матричные методы
Все эти методы используют матрицы межвидовых расстояний <dij> (i, j соответствуют видам), рассчитанные на основе сравнений последовательностей с корректировкой на множественные замещения. Ультраметрические методы (простая иерархическая кластеризация). Достоверны только для стабильных молекулярных часов. В принципе не считаются приемлемыми филогенетическими методами, но могут быть использованы для целей классификации или генерации предварительных филогенетических деревьев.
Метод объединения ближайших соседей (neighbor-joining). Более сложный метод восходящей кластеризации, основанный на минимальном эволюционном критерии (кратчайшая суммарная длина ветвей дерева). Чувствителен к ПДВ и гораздо менее точен, чем метод наибольшего правдоподобия, однако высокоэффективен для вычислений и быстр. Не используется для исчерпывающего филогенетического анализа, но для анализа большого количества последовательностей может быть единственным практически применимым методом.
Метод наименьших квадратов, метод Фитча. Метод измерения расстояний, основанный на минимизации разностей между расстояниями на филогенетическом дереве и в соответствующей матрице расстояний. По точности и эффективности примерно равен методу объединения ближайших соседей. Считается неподходящим для исчерпывающего филогенетического анализа, но используется для построения предварительных филогенетических деревьев для метода наибольшего правдоподобия.
Принцип наибольшей экономии (maximum parsimony)
Не использует матрицы расстояний, вместо этого работает с наборами состояний признаков. Состояниями признаков, в частности, могут быть нуклеотиды или аминокислоты в определенных позициях множественных выравниваний. Принцип наибольшей экономии (НЭ), основанный на принципе наименьшего действия в физике, определяет как наиболее вероятный тот эволюционный сценарий (филогенетическое дерево), который включает в себя наименьшее количество событий (переходов состояний в наборе признаков).
Существует множество алгоритмов, вычисляющих деревья, наиболее соответствующие принципу НЭ и использующие значимые и незначимые признаки. Принцип НЭ часто ставится под сомнение, поскольку существуют деревья, лишь слегка отличающиеся от наиболее экономичного варианта, но имеющие совершенно иную топологию. Метод высоко чувствителен к ПДВ.
Метод наибольшего правдоподобия (maximum likelihood)
Аналогично методу НЭ, в методе наибольшего правдоподобия (НП) оцениваются переходы между состояниями признаков и выбираются деревья, набравшие наибольший вес. В отличие от метода НЭ, метод НП является параметрическим статистическим подходом, который использует детальную модель эволюции признака для оценки вероятности данных на основе имеющегося эволюционного дерева. Дерево, которое имеет наибольшую вероятность возникновения наблюдаемых данных, признается наиболее вероятным. Метод НП зачастую производит деревья, аналогичные тем, которые получаются методом НЭ, но теоретически он предпочтительнее, будучи (в отличие от НЭ) статистически более достоверным (то есть при наличии достаточного количества данных гарантирует получение наиболее правдоподобного дерева). На практике метод НП часто превосходит метод НЭ. Методы НП чрезвычайно затратны с вычислительной точки зрения и непрактичны при работе с большими наборами данных. Таким образом, методы НП зачастую используются для оптимизации предварительных деревьев, полученных методом объединения ближайших соседей и методом Фитча. Для тех же филогенетических исследований, где точность построения дерева важнее скорости, следует выбирать методы НП. Более того, недавние алгоритмические достижения более чем на порядок ускорили построение филогенетических деревьев методами НП без серьезных потерь точности (Price et al., 2010).
Байесовский подход
Подобно методу НП, этот подход использует функцию правдоподобия, но прибегает к теореме Байеса с целью связать апостериорную вероятность дерева с правдоподобием данных и априорную вероятность дерева с эволюционной моделью. В отличие от методов НЭ и НП, которые выводят наилучшее дерево или набор деревьев, методы байесовского вывода выбирают деревья пропорционально их правдоподобию и определяют представительный набор деревьев. Метод хорошо работает для относительно небольших объемов данных, но непрактичен для больших.
Проверка точности филогенетических методов и достоверности деревьев
Модельные деревья
Филогенетические методы постоянно проверяются на искусственно смоделированных данных, для которых известна точная история эволюции. Методы сравниваются по критерию точности реконструкции топологии для искусственно построенных деревьев. Как правило, различные методы НП и байесовские методы превосходят все остальные для небольших наборов данных. Наилучшие результаты показывают итерационные методы, которые используют исходное дерево, построенное по методу НП, чтобы выравнивать данные, перестраивать дерево и повторять так до сходимости.
Бутстреппинг
Наиболее часто используемый тест на надежность топологии филогенетического дерева, при котором рассматриваются выборки данных (колонки выравнивания) и дерево оценивается по большому числу выборок. Процент выборок (то есть репликаций), в которых реконструируется данный узел дерева, называют уровнем поддержки. Статистика бутстреппинга еще не полностью разработана, поэтому пороговые значения для «достаточно высокого» уровня поддержки определяются путем моделирования или эмпирического анализа и могут варьировать в зависимости от целей конкретного исследования (например, значения более 90 процентов, или более 70 процентов; поддержка ниже 50 процентов обычно не считается надежной).
Статистические критерии проверки филогенетических гипотез (топологий деревьев)
Для сравнения правдоподобия различных топологий деревьев, выводимых из одного и того же набора данных, разработаны статистические критерии, основанные на различных моделях правдоподобия (самые известные — критерий Кишино—Хасегавы и приблизительно несмещенный критерий).
Когда исследователь интересуется филогенетическим сродством конкретного таксона, соответствующая ветвь переносится в различные положения в дереве, при сохранении топологии остальных ветвей, и правдоподобие каждого из полученных деревьев сравнивается при помощи статистических критериев с правдоподобием исходного дерева, полученного методом НП. Разновидность этого критерия применяется к деревьям с ограничениями, используемым для проверки филогенетических гипотез, таких как монофилия определенной группы (например, архей) в определенном наборе данных. В этом случае сравнивается правдоподобие дерева с ограничениями (монофилия в данном примере) с правдоподобием исходного НП-дерева.
Часто встречающиеся аномалии филогенетического анализа
Ни один филогенетический метод не застрахован от аномалий, которые часто оказывают заметное влияние на топологию дерева. Двумя основными классами филогенетических аномалий являются гомоплазия и притяжение длинных ветвей (ПДВ). Гомоплазия включает в себя параллельные, сходящиеся и обратные мутации, которые филогенетически не информативны и неверно истолковываются филогенетическими методами. ПДВ называется чрезвычайно распространенный случай, когда длинные ветви (быстро эволюционирующие линии) в дереве кластеризуются вместе только потому, что ни одна из них не проявляет сродства к другим группам, а не потому, что они на самом деле образуют монофилетическую группу. Филогенетики также иногда говорят о притяжении коротких ветвей, то есть ошибочной кластеризации коротких ветвей дерева. Разработка новых методов филогенетического анализа в большой степени побуждается необходимостью преодолеть эти аномалии, сохраняя притом вычислительный метод приемлемым с практической точки зрения.
Общие производные признаки
Важным подходом филогенетического анализа, дополняющим традиционные молекулярные филогенетические методы, является анализ общих производных признаков (так называемых синапоморфий), которые могут быть использованы для разграничения монофилетических групп (клад). Синапоморфии суть признаки, объединяющие всех членов монофилетической группы и исключающие все другие виды. В принципе одна достоверная синапоморфия может определять кладу. Однако это верно только в отсутствие гомоплазии, которую невозможно исключить для большинства признаков. Предполагаемые синапоморфии выбираются таким образом, чтобы свести вероятность гомоплазии к минимуму, например уникальные вставки в консервативных генах, в частности вставки мобильных элементов, мутации, которые требуют нескольких нуклеотидных замен, и слияния генов. В филогеномике идет активный поиск подобных редких геномных изменений. Одних синапоморфий часто недостаточно для несомненных филогенетических выводов, но они предоставляют дополнительные свидетельства для филогений, основанных на геномных последовательностях.
Деревья, не основанные на геномных последовательностях
Филогенетические методы пригодны не только для выравнивания гомологичных последовательностей, но и для анализа дистанционных матриц, полученных полногеномным сравнением любого числа других признаков (таких как содержание общих генов или оперонная организация). Например, в случае содержания общих генов расстояние между двумя геномами определяется как Dij = nij/ni, где nij — число генов, общих для двух геномов, а ni — полное число генов в меньшем геноме. Геномные деревья, полученные этим методом, обычно не являются надежными филогениями из-за обширной гомоплазии. Однако эти деревья могут быть информативными для сравнения образа жизни организмов.
Нейтральная теория молекулярной эволюции
Вероятно, важнейшим прорывом в эволюционной биологии после СТЭ стала нейтральная теория молекулярной эволюции. Как правило, ее связывают с именем Мото Кимуры (Kimura, 1983), хотя Джукс и Кинг одновременно и независимо развивали аналогичные идеи. Вначале нейтральная теория развивалась как логическое продолжение популяционно-генетических идей Райта, основанных на важности генетического дрейфа в эволюции. Согласно нейтральной теории, значительное большинство всех фиксируемых в процессе эволюции мутаций являются относительно нейтральными; таким образом, фиксация возникает на основе случайного дрейфа. Следствием этой теории, неоднократно подчеркиваемым Кимурой, является то, что геномная последовательность эволюционирует равномерно, как по часам (в подтверждение исходной гипотезы молекулярных часов Цукеркандля и Полинга), при этом полезные мутации, подверженные естественному отбору, настолько редки, что ими можно с успехом пренебречь в целях количественного описания эволюционного процесса. Естественно, нейтральная теория отнюдь не подразумевает, что естественный отбор не важен для эволюции. На самом деле теория подчеркивает, что доминирующим способом отбора является не дарвиновский позитивный отбор на основе адаптивных мутаций, а отсекающий (очищающий) отбор, который удаляет вредные мутации, в то же время допуская фиксацию нейтральных мутаций путем генетического дрейфа.
Последующие исследования довели эту теорию до более реалистичной формы: чтобы зафиксироваться, мутация должна быть не в буквальном смысле нейтральной, а всего лишь достаточно мало вредной, чтобы избежать немедленного удаления отсекающим отбором. Современная теория «почти нейтральных» мутаций была разработана в первую очередь Томоко Отой (Ohta, 2002). То, какие мутации распознаются как вредные при вычищении отбором, в большой степени зависит от величины популяции: в небольших популяциях в ходе генного дрейфа могут зафиксироваться даже существенно вредные мутации, тогда как в больших популяциях даже малого негативного эффекта будет достаточно для удаления мутантной аллели (см. табл. 1-1).
Главной эмпирической проверкой теории (почти) нейтральных мутаций является измерение постоянства скорости эволюционного процесса в семействах генов. Несмотря на то что зачастую можно наблюдать значительную дисперсию молекулярных часов, такие измерения с уверенностью показывают, что доля нейтральных мутаций среди зафиксированных и в самом деле весьма существенна (Bromham and Penny, 2003; Novichkov et al., 2004). Теория почти нейтральных мутаций является значительным отступлением от селекционистской парадигмы СТЭ, поскольку однозначно утверждает, что большинство мутаций, зафиксированных в ходе эволюции, не подвержены дарвиновскому (позитивному) отбору. Хотя Дарвин и предвидел нейтралистскую парадигму, утверждая, что для целей классификации лучше всего подходят селективно нейтральные характеристики, однако он не развил эту прозорливую идею, и она, таким образом, не стала частью СТЭ.
Важно отметить, что в ходе последующего развития «нейтральной» теории Кимура, Ота и другие осознали, что те мутации, которые были почти нейтральными во время их фиксации, не были в то же время не важными для эволюции. Напротив, такие мутации составили резервуар вариаций (почти нейтральную сеть аллелей), который может использоваться естественным отбором в свете меняющихся условий среды, — феномен, важный как для микро-, так и для макроэволюции (Kimura, 1991). Эта идея стала ключевой для некоторых позднейших открытий в эволюционной теории, мы обсудим ее более детально позднее в этой книге (в частности, в гл. 8 и 9).
Измерение естественного отбора сравнением последовательностей ДНК
Несмотря на всю свою важность, дарвиновский естественный отбор является концепцией, определенной в качественных терминах. В рамках же популяционной генетики и СТЭ отсекающий и положительный отбор оказались более конкретными и математически определенными. В описании СТЭ отбор скорее можно приравнять к силе в классической механике или потоку в классической термодинамике, то есть к феноменологически определяемому количеству. С появлением сравнения последовательностей ДНК стало возможно обнаруживать и измерять отбор в определенных механистических терминах, базируясь на подсчете различных типов замещений нуклеотидов. Для измерения отбора путем сравнения последовательностей используются две очень простые идеи (см. табл. 2-2). Эти два подхода имеют в своей основе очень много общего, поскольку оба определяют два класса сайтов, один из которых принимается в качестве фона нейтральной эволюции. Первый метод заключается в сравнении числа замен нуклеотидов в позициях, важных с точки зрения кодирования аминокислот (несинонимичные позиции), и в позициях, которые, из-за избыточности генетического кода, не имеют значения для кодирования белков (синонимичные замены). Если отношение скоростей несинонимичных и синонимичных замен (Ka/Ks, см. табл. 2-2) значительно ниже 1, то эволюция соответствующего гена в основном определяется отсекающим отбором, направленным на данную последовательность белка; напротив, в случае Ka/Ks > 1 эволюция определяется в основном положительным дарвиновским отбором (см. табл. 2-2). Второй, более точный подход использует так называемый критерий Макдональда—Крейтмана для измерения отбора, при котором соотношение Ka/Ks сравнивается для внутривидовых вариантов (полиморфизмы) и межвидовых вариантов (фиксированные мутации). Поскольку незафиксированные полиморфизмы в основном нейтральны, то межвидовое отношение Ka/Ks должно быть значительно меньше, чем Ka/Ks для полиморфизмов в случае отсекающего отбора, и значительно больше, чем значение для полиморфизмов в случае положительного отбора.
Таблица 2-2
Измерение отбора путем анализа последовательностей белок-кодирующих генов (Hurst, 2002; Li, 1997)
Белок-кодирующие последовательности состоят из двух видов сайтов:
• синонимичные, в которых замены не влияют на последовательность кодируемых аминокислот;
• несинонимичные, в которых замены ведут к заменам аминокислот.
Отношение Ka/Ks (где Ka — частота несинонимичных замен, Ks — синонимичных; обе вычисляются с коррекцией на множественные замены) является количественной мерой отбора, действующего на уровне белковых последовательностей.
Ka/Ks = 1 — нейтральная эволюция белковой последовательности (кодируемый белок не подвергается отбору).
Для большинства белок-кодирующих генов Ka/Ks ? 1 — отсекающий отбор.
Для прокариот типично Ka/Ks < 0,1.
Для эукариот типично Ka/Ks ? 0,1–0,2.
Ka/Ks > 1 — положительный отбор; достаточно редко встречается для белок-кодирующих генов, но для некоторых категорий генов, несомненно, наличествует, например для генов, участвующих в антипаразитической защите или в сперматогенезе, а также в вирусных белках, таких как гемагглютинин вируса гриппа.
• Для измерения Ka/Ks для индивидуальных сайтов используют методы наибольшего правдоподобия; большинство белоккодирующих генов содержат несколько сайтов, подверженных положительному отбору.
• Использование Ka/Ks для измерения уровня отбора предполагает нейтральность синонимичных сайтов.
• Однако Ka и Ks положительно коррелируют между собой — таким образом, отбор затрагивает и синонимичные сайты.
• Некодирующие сайты, такие как интронные последовательности, могут использоваться как фон нейтральной эволюции при измерении отбора на синонимичных сайтах (Ks/Ki, где Ki — частота замен для интронных сайтов).
• Критерий Макдональда—Крейтмана (Aquadro, 1997; McDonald and Kreitman, 1991) широко используется для измерения отбора. Он сравнивает внутривидовые вариации (частота полиморфизма, P) с межвидовыми вариациями (дивергенция, D).
Dn/Ds = Pn/Ps — нейтральная эволюция белковой последовательности.
Dn/Ds < Pn/Ps — отсекающий отбор.
Dn/Ds > Pn/Ps — положительный отбор.
Появление таких количественных подходов к анализу отбора примечательно не только благодаря их технической применимости в изучении эволюции: они также являются признаком фундаментальных изменений в способах осмысления отбора биологами. Дарвиновская качественная идея, которая была выражена в абстрактной математической форме Фишером и впервые измерена с использованием генетических методов Добржанским и его учениками, теперь превратилась в прямо измеряемую статистическую характеристику ансамблей нуклеотидных сайтов. Такая трансформация концепции отбора сродни тому, как классическая термодинамика с ее абстрактными потоками превратилась в статистическую физику Больцмана и Гиббса (см. гл. 4).
Эгоистичные гены, мусорная ДНК и мобильные элементы
Хоть это и редко утверждается без обиняков, классическая генетика предполагает, что почти все части генома (все нуклеотиды, если употреблять более современные, молекулярные термины) имеют определенные функции. Это неявно выраженное утверждение также важно и с точки зрения СТЭ, с ее панадаптационистским подходом. Однако это понимание подверглось сомнению еще в 1960-х и 1970-х годах по мере накопления данных об отсутствии прямой связи между размером генома и фенотипической сложностью организма. Даже с использованием приблизительных методов, доступных в то время, становилось ясно, что организмы с примерно одинаковым уровнем фенотипической сложности зачастую имеют геномы, на порядок различающиеся по размеру (так называемый парадокс гаплоидной величины). Этот парадокс был концептуально разрешен с помощью двух связанных друг с другом фундаментальных идей: эгоистичных генов и мусорной ДНК[21]. Концепция эгоистичных генов была предложена Ричардом Докинзом в одноименной книге, изданной в 1976 году (Dawkins, 2006). Резко отступив от организм-центричной парадигмы СТЭ, Докинз приходит к выводу, что естественный отбор может действовать не только на уровне организма в целом, но и на уровне индивидуального гена. Этот взгляд, поданный в умышленно провокационной манере, представляет геномы и организмы, по сути, средствами размножения генов.
Концепция эгоистичных генов породила множество важных выводов, и некоторые из них мы рассмотрим ниже в этой книге. Один из аспектов, имеющий непосредственное отношение к парадоксу гаплоидной величины, был всесторонне рассмотрен Фордом Дулиттлом и Кармен Сапиенцей (Doolittle and Sapienza, 1980), а также Лесли Оргелом и Фрэнсисом Криком (Orgel and Crick, 1980). Они предположили, что немалая или даже основная часть геномной ДНК (по крайней мере в сложных многоклеточных организмах) состоит из различных классов повторов, которые образуются в результате амплификации эгоистичных элементов — абсолютных паразитов, говоря хлестким языком Оргела и Крика. Другими словами, с точки зрения организма, большая часть геномной ДНК должна быть признана избыточной. Такой взгляд на геном в корне отличается от панселекционистской парадигмы, присущей СТЭ, в рамках которой большинство или даже все нуклеотиды в геноме подвержены влиянию отсекающего или положительного отбора, действующего на уровне организма.
Концептуально родственным важным открытием стало обнаружение транспозонов, или «прыгающих генов», сначала Барбарой Макклинток в 1940-х годах в растениях, а затем и в животных. Эти транспозоны стали затем известны как мобильные элементы (то есть генетические элементы, которые имеют тенденцию часто менять свое место в геноме; McClintock, 1984). Демонстрация вездесущности мобильных элементов привела к концепции высокодинамичных, постоянно меняющихся геномов задолго до рождения современной геномики[22].
Эволюция путем дупликации генов и геномов: ортологи и паралоги
СТЭ в полной мере унаследовала центральное положение дарвиновской теории, провозгласившей постепенные малые изменения единственно возможным материалом для эволюции. Однако эта концепция была поставлена под сомнение альтернативной концепцией эволюции дупликацией гена, разработанной Сусуму Оно в его классической книге 1970 года (Ohno, 1970). Мысль о том, что дупликация частей хромосом может служить одной из движущих сил эволюции, восходит к основателям современной количественной генетики, в частности к Фишеру и Холдейну[23]. Однако Оно первым предположил, что дупликация генов является основой эволюции геномов и организмов, и первым подвел качественную теорию под это положение. Начав с цитогенетических свидетельств полногеномной дупликации (ПГД) в начале эволюции хордовых, Оно выдвинул гипотезу о том, что дупликация генов является важным, если не единственным, путем эволюционии новых биологических функций. Согласно гипотезе Оно, дупликация гена высвобождает одну из копий от ограничений отсекающего отбора и, таким образом, эта копия получает потенциал развития новой функции (феномен, позднее названный неофункционализацией). Очевидно, что возникновение нового гена в результате дупликации, не говоря уже о дупликации геномного участка или ПГД, является огромным отличием от дарвиновских ничтожно малых изменений. Если такие крупные события в самом деле являются ключевыми для эволюции, то парадигма постепенных изменений в опасности. Позднейшие исследования дупликации генов, обсуждаемые далее в этой книге (см. гл. 8 и 9), привели к предположению о том, что неофункционализация вряд ли является основным путем эволюции дуплицированных генов. Однако факт остается фактом: дупликация, как важнейший механизм эволюции, бросает вызов градуализму.
Примерно в то же время, когда была издана книга Оно об эволюции путем дупликации генов, Уолтер Фитч опубликовал весьма плодотворную статью, всю значимость которой стало возможным оценить лишь в свете более поздних достижений геномики. Фитч исследовал понятие гомологии (общего предка) генов и провел различие между двумя классами гомологичных генов: ортологами и паралогами (Fitch, 1970). Ортологи — это гены, которые эволюционировали вертикально от одного предкового гена, принадлежащего общему предку сравниваемых организмов, тогда как паралоги — гены, эволюционировавшие в результате дупликации. Понятия ортологии и паралогии очевидным образом тесно связаны между собой и зависят от конкретной топологии филогенетического дерева данного семейства генов, так что дупликация в определенном узле дерева порождает новый набор паралогов в поддереве-потомке (подробнее см. в гл. 3). Более того, концептуальное определение паралогии осложнено специфичными для каждой линии эволюции потерей и горизонтальным переносом генов (см. гл. 5 и 7). Тем не менее, если не принимать во внимание эти осложнения, классификация гомологов Фитча остается центральной для эволюционной геномики[24].
Прерывистое равновесие и несостоятельность градуализма
Недостача межвидовых переходных форм в палеонтологической летописи — постоянная тема эволюционной биологии. Дарвин осознавал эту проблему и традиционно считал ее (так же как и палеонтологи, следующие дарвиновским традициям) следствием драматической неполноты этой летописи. Однако обширное накопление палеонтологических данных в XX веке мало помогло (если не сказать совершенно не помогло) в решении этой проблемы, что привело к возникновению иной точки зрения, сначала с появлением концепции квантовой эволюции Джорджа Гэйлорда Симпсона, затем оформившейся в концепцию прерывистого равновесия Стивена Джея Гулда и Нильса Элдриджа (Eldredge and Gould, 1997; Gould, 2002). Гулд и Элдридж собрали обширную доказательную базу, свидетельствующую о том, что история большинства видов животных, отраженная в палеонтологической летописи, соответствует в основном состоянию покоя — то есть, фактически, отсутствия изменений. Состояние покоя (стасис) перемежается «внезапным» исчезновением видов, последовательно замещаемых новыми. Следствием такой модели является очень быстрое в сравнении с продолжительностью стасиса видообразование; возникновение новых видов в определенной области является следствием миграции из области видообразования; градуалистское видообразование (постепенная трансформация видов в новые) — довольно редкий процесс. Такая модель прерывистого равновесия кажется применимой и к эволюции высших таксонов и зачастую обобщается до несостоятельности градуализма в целом, хотя правомерность такого вывода часто подвергается критике.
Пандативы, экзаптация, эволюция как ремесленник и ошибочность панглоссианской парадигмы эволюции
Пусть и неявно, но принципу градуализма был брошен вызов гипотезой Оно об эволюции генов и геномов путем дупликации, a затем, в явной форме, концепцией прерывистого равновесия. Адаптационистская программа эволюционной биологии подверглась решительной, сметающей все на своем пути атаке в статье 1979 года «Пандативы Святого Марка» Гулда и Левонтина (Gould and Lewontin, 1979), одной из самых необычных и влиятельных статей в истории биологии. Гулд и Левонтин саркастически описали адаптационистскую картину мира как панглоссианскую парадигму, названную так в честь примечательного персонажа вольтеровского «Кандида», который утверждал, что «все к лучшему в этом мире» [пер. Ф. Сологуба] (даже катастрофы). Гулд и Левонтин подчеркивали, что вместо того, чтобы стряпать на скорую руку «сказки просто так»[25] о правдоподобных адаптациях, эволюционным биологам следовало бы искать объяснение наблюдаемых черт организации биологических организмов исходя из плюралистского подхода, который принимает во внимание не только отбор, но также и внутренние ограничения, случайный дрейф и другие факторы. Метафора пандатива означает, что многие функционально важные элементы биологической организации вовсе не эволюционировали как специальные устройства для выполнения определенных функций, но скорее являются продуктами неадаптивных архитектурных ограничений, подобно пандативам (spandrels), появляющимся в арках соборов и других зданий исключительно вследствие требований конструкции, и могут использоваться для различных целей, например для украшения собора (см. рис. 2-2). Процессу использования пандативов в биологии было дано специальное название экзаптация, и Гулд провозгласил его важным путем эволюции (Gould, 1997a). Концепция пандативов связана с почти нейтральной теорией, но в каком-то смысле идет дальше и подходит ближе к сути эволюционного мышления, показывая, что даже те фенотипические черты, которые выглядят как типичные адаптации, не обязательно эволюционировали под давлением естественного отбора.

Рис. 2-2. Один из пандативов базилики Святого Марка в Венеции. Фото Марии Шнитцмейер, Викисклад.
В более ранней статье по сходной тематике Франсуа Жакоб (один из первооткрывателей регуляции генов и автор нескольких других плодотворных идей в бактериальной генетике, см. гл. 5) ввел метафору мастера-самоучки. Отталкиваясь прежде всего от сравнительного анализа механизмов развития, Жакоб положил в основу своих рассуждений, что эволюция действует не как инженер или дизайнер, а скорее как ремесленник-самоучка, причем чрезвычайно зависимый от предыдущего опыта при решении стоящих перед ним проблем:
«Сложно проводить аналогии между естественным отбором и какими-то аспектами человеческого поведения. Однако если очень хочется поиграть в сравнения, то можно сказать, что естественный отбор работает не как изобретатель или инженер. Он работает как дилетант — мастер на все руки, который не знает точно, что он собирается создать, и при этом использует все, что подвернется под руку, будь то обрывки ниток, куски дерева или старые коробки; короче, он действует как тот мастеровой, который использует все, что есть в его распоряжении, чтобы сделать хоть что-то, лишь бы работало» (Jacob, 1977).
Ключевым выводом концепции ремесленника-самоучки становится то, что итоговый результат эволюции непредсказуем, или по крайней мере его невозможно предсказать, не зная в деталях всех предшествующих событий. Другими словами, если взять и «проиграть эволюционную пластинку заново» (любимая метафора Гулда) в некоем мысленном эксперименте, то результат будет отличен от того, что мы наблюдаем в реальности, возможно до неузнаваемости; мы вернемся к этому обсуждению позднее в этой книге (см. гл. 13).
Эволюция в мире микробов и вирусов и трехдоменное древо жизни
Вероятно, в ходе развития биологии наибольшее влияние на изменение представления об эволюции оказало распространение эволюционных исследований на мир микробов, а именно одноклеточных эукариот (протист), прокариот (бактерий и архей) и вирусов. Дарвиновское представление об эволюции и все достижения эволюционной биологии нескольких последующих десятилетий базировались исключительно на исследовании животных и растений, тогда как одноклеточные эукариоты (протисты) и бактерии (монеры) были сугубо номинально размещены у корня древа жизни Эрнстом Геккелем и его последователями. Хотя к 1950-м годам генетический анализ бактериофагов и бактерий продвинулся настолько, что стало очевидным, что эти формы жизни обладают эволюционирующими геномами, СТЭ не принимала во внимание эти открытия. То, что бактерии (не говоря уже о вирусах) эволюционируют по тому же самому принципу и с использованием тех же механизмов, что и животные и растения, отнюдь не очевидно, учитывая все их разительные биологические отличия от многоклеточных организмов, и в особенности из-за отсутствия у них типичного полового размножения и репродуктивной изоляции, ключевых для видообразования среди животных и растений.
Фактически прокариоты стали «видны» эволюционным биологам в 1977 году, после выхода революционной работы Вёзе и его коллег по филогенезу рРНК (Woese, 1987)[26]. Рассмотренное в общем контексте, открытие Вёзе является эпохально важным и, возможно, даже заслуживает сравнения с открытием структуры ДНК. Вёзе установил, что в одной молекулярной структуре, а именно последовательности нуклеотидов рРНК, выявляется очевидная консервативность во всем диапазоне клеточных форм жизни. Кроме того, чрезвычайно информативным оказался и филогенетический анализ этой универсальной консервативной молекулы: он показал, что рРНК, в некотором приближении, эволюционирует с постоянной скоростью, то есть подчиняется модели молекулярных часов. Это привело к еще одному важному открытию, ставшему одним из символов эволюционной биологии конца XX века, — трехдоменному древу жизни (см. рис. 2-3; Woese et al., 1990). Тремя доменами являются бактерии, археи и эукариоты. Домен архей был открыт Джорджем Фоксом и Вёзе сравнительным анализом рРНК, когда в новой группе ничем, казалось бы, не примечательных «бактерий» обнаружились существенные отличия как от остальных бактерий, так и от более сложных эукариотических организмов. В дополнение к разграничению трех доменов, Вёзе и его коллеги использовали филогенетический анализ рРНК для идентификации нескольких основных ветвей архей и бактерий (Woese, 1987). Из этого следовало, что эволюция прокариот столь же доступна для изучения, как эволюция сложных эукариот, — концепция, чуждая микробиологам до работы Вёзе (Stanier and Van Niel, 1962). Благодаря достижениям Вёзе, его сотрудников и последователей появилась все усиливающаяся тенденция приравнивать филогенетическое древо рРНК, с его трехдоменной структурой, к древу жизни Дарвина и Геккеля (Pace, 2009a, 2006). В течение нескольких лет после публикации открытий Вёзе стало ясно, что топологически древо рРНК (по крайней мере, в своих основных чертах) конгруэнтно деревьям некоторых из самых консервативных белков, таких как рибосомные белки, факторы трансляции, субъ единицы ДНК-зависимой РНК-полимеразы и мембранные АТФазы.
Более 800 000 книг и аудиокниг! 📚
Получи 2 месяца Литрес Подписки в подарок и наслаждайся неограниченным чтением
ПОЛУЧИТЬ ПОДАРОКДанный текст является ознакомительным фрагментом.
Читайте также
Пути эволюции предопределены на молекулярном уровне
Пути эволюции предопределены на молекулярном уровне В наши дни бурное развитие молекулярной биологии привело к тому, что многие важные биологические закономерности, в том числе явление параллельной эволюции, временно оказались как бы за рамками «настоящей серьезной
2.1. Становление и основные положения синтетической теории эволюции
2.1. Становление и основные положения синтетической теории эволюции Эволюционизм возник как альтернатива учению о неизменности видов. Вопросы, связанные с возникновением и развитием жизни, прошли через всю интеллектуальную историю человечества. Количество литературы,
2.2. Альтернативные теории эволюции
2.2. Альтернативные теории эволюции Многообразие альтернативных концепций эволюции обычно группируют в три ветви: ламаркизм, теории направленной эволюции и сальтационизм. Каждая ветвь имеет свою богатую историю. В настоящее время эти названия представляют скорее
Глава 1 Основы эволюции: Дарвин и синтетическая теория эволюции
Глава 1 Основы эволюции: Дарвин и синтетическая теория эволюции Пер. А. НадирянВ этой и следующей главах дается краткое описание современного состояния эволюционной биологии, какой она была до 1995 года, когда возникло новое направление науки — сравнительная геномика.
Глава 9 Ламарковский, дарвиновский и райтовский режимы эволюции, эволюция эволюционируемости, надежность биологических систем и созидательная роль шума в эволюции
Глава 9 Ламарковский, дарвиновский и райтовский режимы эволюции, эволюция эволюционируемости, надежность биологических систем и созидательная роль шума в эволюции Пер. Д. ТулиноваДрама ламаркизмаКак уже отмечалось в предисловии к данной книге, одной из ключевых заслуг
Глава I ОБЩИЕ ЗАМЕЧАНИЯ О ТЕОРИИ ЭВОЛЮЦИИ
Глава I ОБЩИЕ ЗАМЕЧАНИЯ О ТЕОРИИ ЭВОЛЮЦИИ …Дальнейшее исследование должно весьма значительно модифицировать нынешние, в том числе и строго дарвинистские, представления о процессе развития видов. Ф. Энгельс.
НАУЧНОСТЬ ТЕОРИИ ЭВОЛЮЦИИ
НАУЧНОСТЬ ТЕОРИИ ЭВОЛЮЦИИ Каждому, вероятно, приходилось слышать время от времени, что в биологии нет настоящих теорий. В частности, эволюционизму отказывают в статусе подлинной научной теории по следующим соображениям.1. Это в основном описание всевозможных событий, а
КРИТИКА СИНТЕТИЧЕСКОЙ ТЕОРИИ ЭВОЛЮЦИИ
КРИТИКА СИНТЕТИЧЕСКОЙ ТЕОРИИ ЭВОЛЮЦИИ Не считая критику синтетической теории эволюции (СТЭ) специальной задачей, я должен тем не менее пояснить свое отношение к господствующим сейчас взглядам, иначе трудно рассчитывать на сочувствие читателя к попытке изменить их. Ниже
6.3. Функциональные блоки и механизмы эволюции
6.3. Функциональные блоки и механизмы эволюции Концепция универсальных функциональных блоков дает возможность проанализировать некоторые стороны эволюционного процесса, так как функциональный блок является тем элементом, из которого построена вся система функций
2.5. Возможные пути предклеточной эволюции
2.5. Возможные пути предклеточной эволюции Было бы большим упрощением полагать, что описанная выше химическая эволюция, в ходе которой накапливались все более сложные органические соединения, непосредственно предшествовала клеточной эволюции, т. е. появлению жизни. На
Глава 1. Теории прогрессивной эволюции
Глава 1. Теории прогрессивной эволюции Кто знает, что дух человека возносится ввысь, А дух скота — тот вниз уходит в землю? Экклесиаст, III, 21* Бытовой антропоцентризмЧеловек может оставаться равнодушным ко многому, только не к собственной персоне. В себе его интересует
Половое размножение: пути эволюции
Половое размножение: пути эволюции Размножение половым путем возникло в процессе эволюции не сразу. Первые простейшие одноклеточные существа типа амеб, жгутиконосцев (эвглена зеленая), инфузорий (инфузория-туфелька), радиолярий (солнечник) размножались простым делением