Глава 3 Сравнительная геномика: эволюционирующие геномные ландшафты
Глава 3
Сравнительная геномика: эволюционирующие геномные ландшафты
Пер. А. Неизвестного
Важность перехода к геномике
В догеномную эру были установлены фундаментальные принципы молекулярной эволюции и было сделано немало конкретных наблюдений, имеющих большое значение и повлиявших на основы эволюционной биологии (см. гл. 1 и 2). Но масштабные работы по расшифровке геномов, начавшиеся в середине 90-х и стремительно развивавшиеся в новом тысячелетии, качественно изменили всю эволюционную биологию. Важность обширной базы данных геномных последовательностей, имеющих различную степень расхождения, очевидна. Этот материал дает исследователям возможность изучать механизмы и отдельные события эволюции с необходимой статистической точностью и выявлять даже самые малозаметные эволюционные подвижки. Как бы то ни было, в эволюционной биологии получение разнообразных и полных геномных последовательностей чрезвычайно важно далеко не только ради накопления количества данных. Действительно, лишь полностью расшифрованный геном (в отличие от, скажем, расшифрованного лишь на 95 процентов) дает исследователю объективное и непредвзятое представление о генном репертуаре той или иной формы жизни. Иными словами, исследователь может определить присутствие в организме тех или иных генов и, что одинаково важно, их отсутствие. Таким образом, сравнение полных геномов представляет собой единственный удовлетворяющий исследователя путь к реконструкции эволюции. Открывающаяся картина во многом отличается от всего, что можно было себе представить, оставаясь в рамках традиционной эволюционной биологии.
Если мы действительно стремимся «понять» эволюцию, принципиально важно исследовать геномные образцы как вглубь (для этого необходимы геномные последовательности множества близкородственных представителей одного и того же таксона), так и вширь (для этой цели нужны последовательности как можно большего числа различных таксонов — в идеале всех таксонов). Ко времени написания этих строк, в последние дни 2010 года, собрание секвенированных геномов состояло из нескольких тысяч геномов вирусов, более чем тысячи геномов бактерий и архей, а также приблизительно сотни геномов эукариот. Ко времени издания этой книги геномная база данных почти удвоится, а благодаря новому поколению методов секвенирования в предстоящие годы ее темпы роста должны еще более ускориться[30]. Несмотря на то что не все основные таксоны должным образом охвачены, быстро пополняющееся собрание геномов все более отвечает потребностям исследований как в области микроэволюции, так и в области макроэволюции.
Успехи традиционной геномики дополняют и стремительно накапливающиеся в последнее время, обширные по объему данные по метагеномике — а именно всеобъемлющее (или, по меньшей мере, обширное) секвенирование нуклеиновых кислот форм жизни из разнообразных сред обитания. Хотя применяемые в настоящее время в метагеномике подходы обычно не обеспечивают полную расшифровку геномов, они предоставляют бесценную, объективную информацию о разнообразии жизни в различных средах.
В данной главе представлен обзор разнообразия и основных характеристик геномов. В последующих главах подробно исследуется влияние результатов сравнительных геномных исследований на развитие «постсовременной» синтетической теории эволюционной биологии.
Эволюция геномных ландшафтов
Поразительное разнообразие геномов
Геном стал первым термином с окончанием «-ом» — и до сих пор является наиболее употребительным термином этой группы[31]. Как это всегда бывает в биологии, определить, что же такое геном, нелегко. Говоря просто, геном — это генетическая информация конкретного организма во всей ее полноте. Существование стабильного ядра унаследованной генетической информации (а более конкретно, генов) вытекает из самого факта существования надежной наследственности, а в терминах более фундаментальных — из принципа подверженной ошибкам репликации (ПОР, см. гл. 2). Однако связь между «генетической информацией во всей ее полноте» и «стабильным ядром» не так уж проста. Стоит, к примеру, задать на первый взгляд невинный вопрос: «Что есть геном кишечной палочки Escherichia coli?» — как тут же возникает целый ряд серьезных затруднений. А вопрос «Что такое геном человека?» вызывает свои, не менее сложные проблемы. Вернемся мы к этому обсуждению позднее (см. гл. 5), а сейчас рассмотрим многообразие геномов, расшифрованных за последние 15 лет.
Новая эра геномики наступила на исходе лета 1995 года. Тогда лаборатория Дж. Крейга Вентера опубликовала результаты секвенирования генома условно-патогенной бактерии гемофильного гриппа Haemophilus influenzae (Fleischmann et al., 1995). В процессе расшифровки геномной последовательности H. influenzae Вентер, Гамильтон Смит и их коллеги усовершенствовали так называемый «метод дробовика». Этот подход грубого деления генома на короткие произвольные участки с расшифровкой их по частям и последующим восстановлением полной геномной последовательности быстро превратил секвенирование длинных нуклеотидных цепочек в рутинное дело. В течение года были расшифрованы геномы некоторых других бактерий, первый геном археи (Methanocaldococcus jannaschii) и первый геном эукариота (пекарские дрожжи Saccharomyces cerevisiae) (Koonin et al., 1996). К 1999 году установился стабильный экспоненциальный рост коллекции секвенированных геномов (см. рис. 3-1).
В диапазоне от вирусов до животных геномы различаются по размеру на шесть порядков — от нескольких тысяч до нескольких миллиардов нуклеотидов; для клеточных организмов, исключая вирусы, ширина диапазона составляет четыре порядка (см. рис. 3-2). По количеству генов диапазон значительно уже и составляет всего около четырех порядков, от двух-трех генов у простейших вирусов до приблизительно 40 тысяч генов у некоторых животных. Если же исключить вирусы и паразитические (симбиотические) бактерии, диапазон по числу генов становится довольно узким, немногим более одного порядка (см. рис. 3-2; Koonin, 2009a; Lynch, 2007c). Кажется весьма удивительным, что млекопитающие или цветковые растения имеют всего примерно в десять раз больше (легко идентифицируемых) генов, чем какая-нибудь средняя свободно живущая бактерия, и лишь примерно в два раза больше, чем бактерия из разряда наиболее сложных (см. рис. 3-2). Далее в книге рассматриваются всевозможные объяснения этих явных ограничений по числу генов в геномах всех форм жизни (см. гл. 5, 7 и 10).
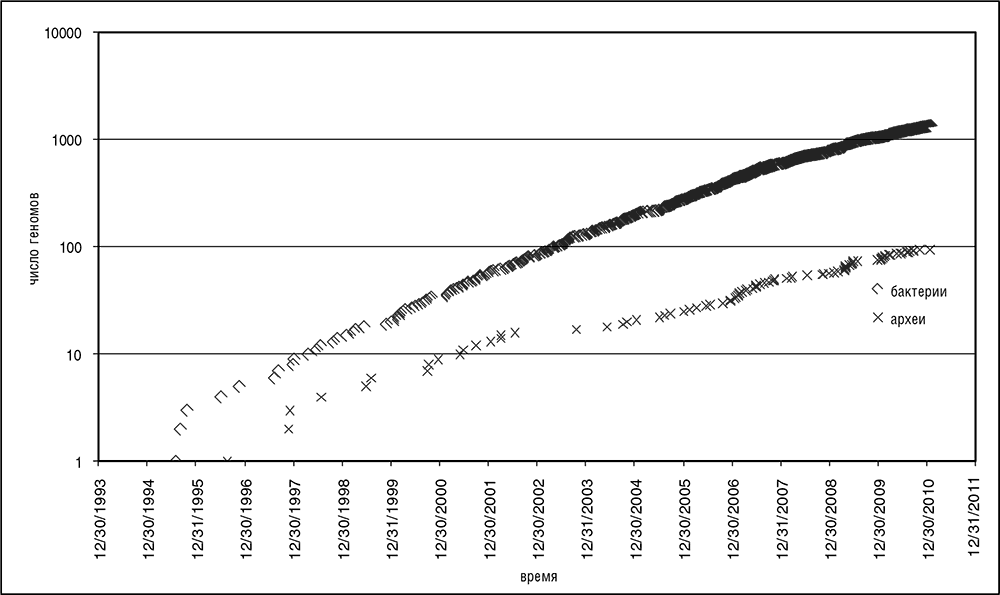
Рис. 3-1. Экспоненциальный рост коллекции секвенированных геномов. Данные с веб-сайта Национального центра биотехнологической информации (www.ncbi.nlm.nih.gov/genome/)
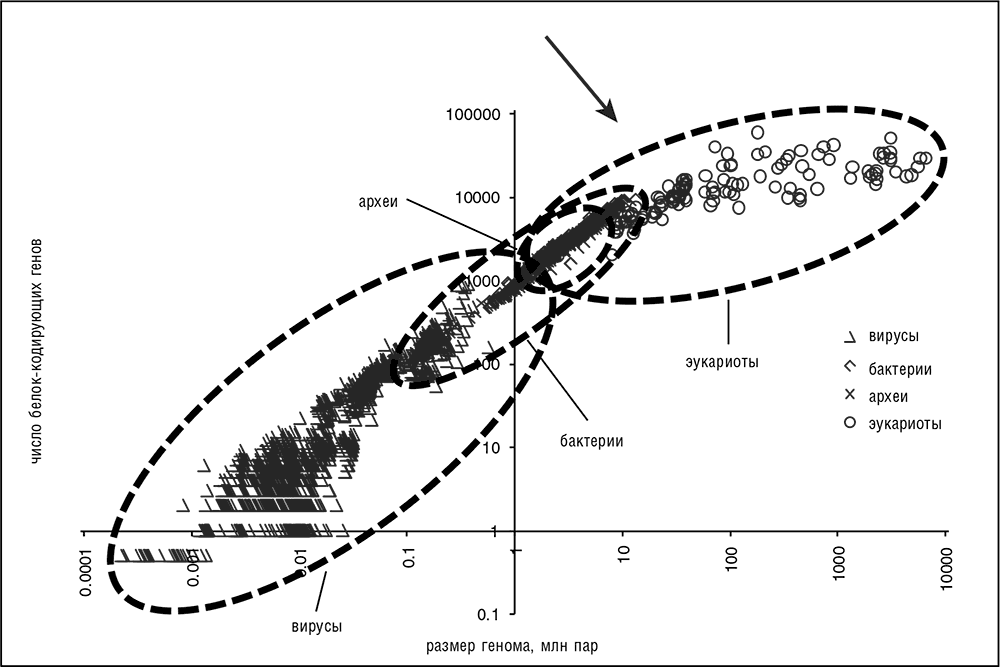
Рис. 3-2. Общий размер геномов и число генов у вирусов, бактерий, архей и эукариот. Данные с веб-сайта Национального центра биотехнологической информации. Представлено в двойном логарифмическом масштабе. Стрелка указывает на точку изменения наклона кривой, соответствующую переходу от «малых» к «большим» геномам.
Грубо говоря, геномы могут быть разделены на два четко выделенных класса (Koonin, 2009a). Граница, разделяющая эти классы, находится в точке изменения наклона кривой на графике, представленном на рис. 3-2.
1. Геномы со строгим соответствием между размером генома и числом генов. К ним относятся геномы всех вирусов и прокариот, имеющие огромную плотность генов от 0,5 до 2 генов на тысячу пар оснований и очень короткие участки между генами (10–15 процентов длины генома и даже меньше), состоящие главным образом из регуляторных элементов. Иногда, говоря о таких геномах, вспоминают ковер «от стены к стене» (wall to wall genomes)[32], так как они почти полностью состоят из легко определяемых генов. Геномы большинства одноклеточных эукариот демонстрируют несколько меньшую зависимость между размером генома и числом генов, чем геномы вирусов и прокариот, тем не менее они могут быть отнесены к этому же классу.
2. Геномы, у которых нет четкой взаимосвязи между размером генома и числом генов, в частности большие геномы многоклеточных и некоторых одноклеточных эукариот. Здесь в лучшем случае наблюдается слабая корреляция между общим размером генома и числом генов. Соответственно, доля генома, занимаемая межгенными участками (а также другими некодирующими последовательностями, такими как интроны), сильно варьирует. В некоторых наиболее сложных геномах, в частности у млекопитающих, основную часть генома составляют именно некодирующие последовательности.
Вариабельность размеров генома и числа генов дополняется разнообразием в других измерениях — например, в физической организации и композиции нуклеотидов. При рассмотрении как вирусной, так и клеточной формы жизни геномы предстают во всевозможных формах нуклеиновых кислот (подробнее см. в гл. 10). Все геномы клеточных организмов состоят из двухцепочечных ДНК, однако количество геномных сегментов (хромосом) и их размеры, форма (кольцевая или линейная), а также плоидность (число наборов) широко разнятся. Азбучная истина гласит, что прокариоты имеют гаплоидные, простые кольцевые хромосомы, в то время как у геномов эукариот, сильно различающихся по плоидности, гены распределены между множеством линейных хромосом. И хотя такие геномные формы, по-видимому, действительно доминируют, на самом деле разнообразие геномов выходит далеко за рамки такого простого дихотомического разделения. В частности, у многих прокариот имеется несколько хромосом, в отдельных случаях — линейных. Более того, вопреки распространенному заблуждению, у прокариот большинство клеток не гаплоидные, то есть они содержат несколько копий генома.
Древние гены составляют в геноме большинство и имеют отчетливую эволюционную судьбу
Как уже упоминалось в предыдущей главе, Эрнст Майр, великий эволюционист XX века и один из основателей СТЭ, с уверенностью предсказывал исходя из больших фенотипических различий между организмами, что гены разных организмов, даже близкородственных, не будут иметь узнаваемого сходства. Ошибочность этого предсказания оказалась просто феерической, что само по себе делает его нетривиальным и ценным. Сравнение последовательностей даже в догеномный период выявило высокий консерватизм последовательностей у некоторых гомологичных белков и молекул некодирующих РНК по всему спектру жизни, от бактерий до млекопитающих (см. предыдущую главу). Более того, высокая степень сходства последовательностей существует у древних паралогов, которые, по-видимому, происходят от копий, ведущих свое происхождение от LUCA (Gogarten et al., 1989; Iwabe et al., 1989). Геномика позволяет перевести это общее понимание в количественное разбиение генов любого генома на классы эволюционной консервативности (см. рис. 3-3; Koonin and Wolf, 2008b).
Ключевое открытие сравнительной геномики состоит в том, что большинство генов в каждом геноме могут считаться высококонсервативными — они имеют легко обнаруживаемые гомологи в организмах, разделяемых сотнями миллионов лет эволюции (например, в случае генов человека, на уровне общего предка позвоночных; см. рис. 3-3; Wolf et al., 2009). Это открытие демонстрирует поразительную устойчивость последовательностей РНК и белков в процессе эволюции: типичное время исчезновения сходства последовательностей у гомологичных генов сравнимо со временем существования жизни на Земле. Помимо основополагающего значения, данный факт имеет огромные практические последствия: благодаря ему, прежде всего, сравнительная геномика становится крайне информативной и действенной.
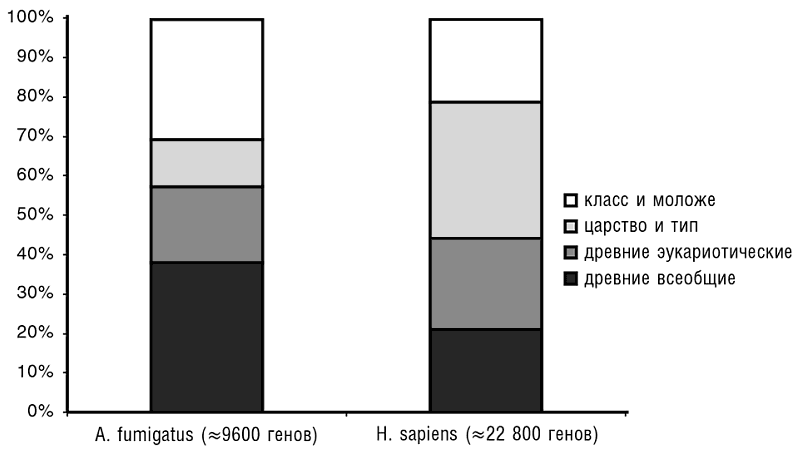
Рис. 3-3. Распределение генов по эволюционному возрасту. «Эволюционный возраст» соответствует самому старому таксономическому узлу, в котором могут быть определены гомологи для белка, производимого данным геном. В частности, для человека древние всеобщие означает «гомологи, обнаруживаемые у прокариот», древние эукариотические означает «гомологи, обнаруживаемые у прокариот вне супергруппы униконтов» (см. гл. 7), царство и тип означает «гомологи, обнаруживаемые у животных вне класса млекопитающих», а класс и моложе означает «вне класса млекопитающих гомологи не обнаружены» (данные по Wolf et al., 2009)
Структуру эволюционного процесса определяют не только консервативные последовательности. На протяжении чрезвычайно длительных эволюционных периодов не просто сохраняется сходство последовательностей РНК и белков, но и гены имеют свойство сохранять свою уникальность. Иными словами, большинство генов развиваются как ортологичные линии, с редкими случаями дупликации (Koonin, 2005). Устойчивость ортологии генов становится очевидной благодаря простой процедуре, широко применяемой в сравнительной геномике и позволяющей эффективно выявлять ортологичные наборы генов. При этом ортологи обнаруживаются как «наилучшие совпадения при двунаправленном сравнении» (bidirectional best hits): все закодированные в геноме белковые последовательности сравниваются со всеми белками, закодированными в другом геноме, a затем процедура повторяется в обратном направлении (Tatusov et al., 1997). Пары генов, дающие наилучшие совпадения (те, которые демонстрируют наибольшее сходство последовательностей) при обоих направлениях сравнения, считаются возможными ортологами; нетрудно применить эту процедуру к нескольким видам путем совмещения треугольников двунаправленных совпадений, имеющих общую сторону (см. табл. 3-1). Примечательно, что такой прямолинейный подход в большинстве случаев хорошо срабатывает: к примеру, порядка 70 процентов генов организмов, разделенных приблизительно 100 миллионами лет эволюции, таких как люди и мыши, легко идентифицируются как ортологи при помощи описанной процедуры (Wolf et al., 2009). Если применить простую модификацию этого алгоритма и включить дупликации генов, характерных для одной линии наследования (дупликации, образовавшиеся после расхождения сравниваемых видов), такой подход позволяет идентифицировать наборы ортологов (известных как кластеры ортологичных генов, КОГ) во многих геномах, в том числе столь удаленных друг от друга, как археи и бактерии — представители двух доменов прокариот (см. гл. 5). Более точные и мощные способы обнаружения ортологов требуют подробного анализа филогенетических деревьев (см. табл. 3-1); впрочем, результаты такого анализа обычно близки к тем, что дают более простые методы, основанные только на сравнении последовательностей. Разумеется, для части генов история дупликаций и потерь настолько сложна, что обнаружить КОГ трудно, поэтому они становятся нечеткими кластерами с неопределенной внутренней структурой. По счастью, этих «трудных» генов в каждом геноме относительно немного.
Таблица 3-1
Классификация гомологичных связей генов: ортологи, паралоги и методы их определения
Эволюционные связи генов:
• Гомология: гены, имеющие общее происхождение.
• Ортология: гомологичные гены, эволюционировавшие путем видообразования.
• Паралогия: гомологичные гены, эволюционировавшие путем дупликации.
• Ксенология: гомологичные гены, имитирующие ортологи, но образовавшиеся в результате горизонтального переноса гена из другой ветви.
• Паралогия, внутренняя и внешняя: паралогичные гены, возникшие в результате видоспецифической дупликации после (внутренняя) или до (внешняя) определенного события видообразования.
• Со-ортология: внутренне-паралогичные гены, совокупно ортологичные по отношению к генам другой ветви (из-за их общего происхождения в ходе видообразования).
• Ортологичная группа (КОГ): совокупность всех потомков данного предкового гена.
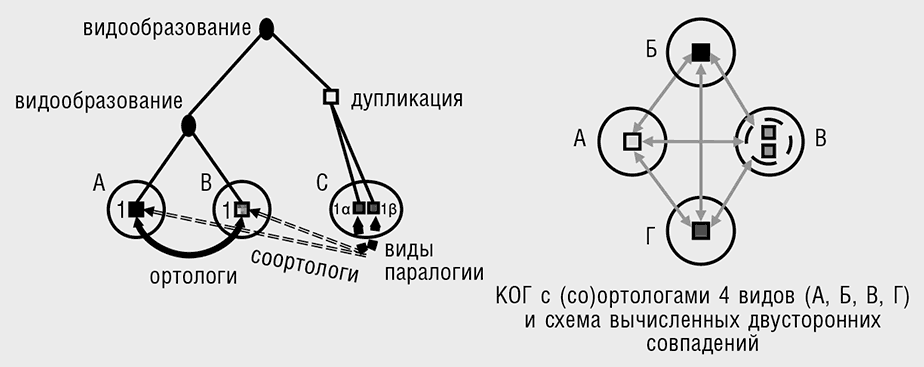
Изначально не вполне складная аббревиатура КОГ относилась к кластерам ортологичных групп (белков), чтобы обозначать соортологичные связи, вызванные дупликацией генов (см. табл. 3-1; Tatusov et al., 1997). Сейчас я предпочитаю расшифровывать КОГ просто как кластеры ортологичных генов, однако само по себе это сокращение остается чрезвычайно удобным для обозначения фундаментального свойства таких кластеров. Эта трехбуквенная аббревиатура широко используется в литературе, и я использую ее в данной книге в качестве сокращенного названия наборов ортологичных генов. Обычно каждый секвенированный геном более чем на 70 процентов состоит из генов, относящихся к КОГ (см. рис. 3-4). В эволюции генома, к которой мы обращаемся в этой книге неоднократно, эта величина представляется важной. Таким образом, существенное большинство генов в каждом геноме весьма консервативно, то есть представлено ортологами во многих далеко отстоящих друг от друга организмах.
Мультидоменные белки и сложность связей ортологов
В этой главе основной упор делается на рассмотрении взаимосвязи между стабильностью и изменчивостью в ходе эволюции. В настоящем разделе мы сосредоточим внимание на отдельных элементах белковой структуры, доменах и мультидоменной организации многих белков (Doolittle, 1995). Таким образом, мы заглянем по другую сторону генной эволюции, которая противостоит стабильности ортологичных линий, отмеченной ранее, и дополняет ее. Домен — центральное понятие в исследовании белков, и определение ему можно дать по меньшей мере на двух уровнях. По первому определению, домены представляют собой компактные элементы белковой структуры с характерными размерами около ста аминокислотных остатков. В этой главе нас интересуют родственные связи геномов, в частности ортология, поэтому необходимости рассматривать структурные элементы нет. Второе определение доменов относится к компактным единицам эволюции, которые могут охватывать один или несколько структурных элементов; здесь нас интересуют именно такие эволюционные домены.
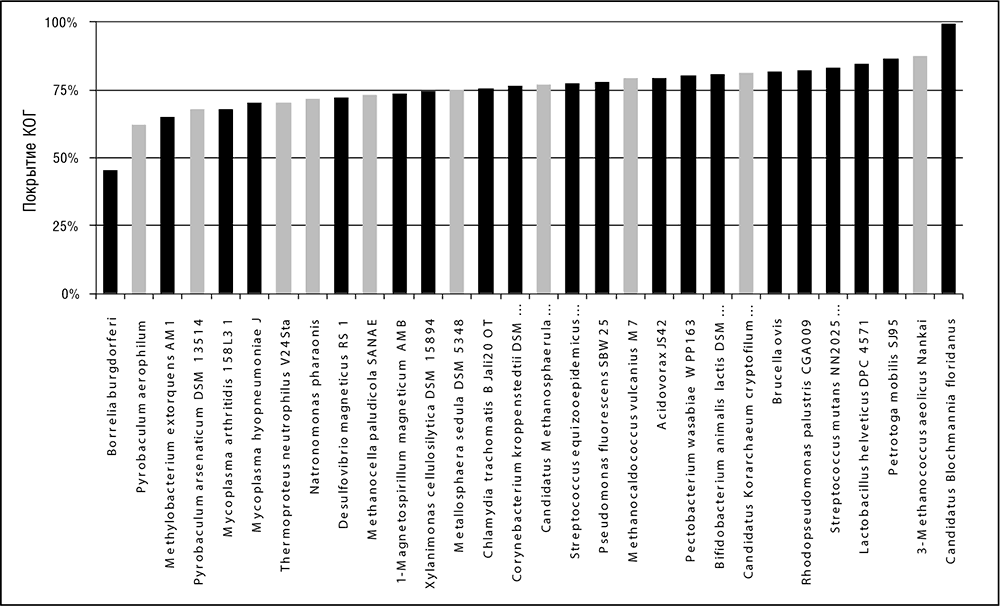
Рис. 3-4. Уровень покрытия КОГ в геномах архей и бактерий. Полные наборы белков в 20 отобранных геномах бактерий (показаны черным) и 10 геномах архей (показаны серым), отнесенные к КОГ (Tatusov et al., 2003). Применялся метод COGNITOR (Makarova et al., 2007b)
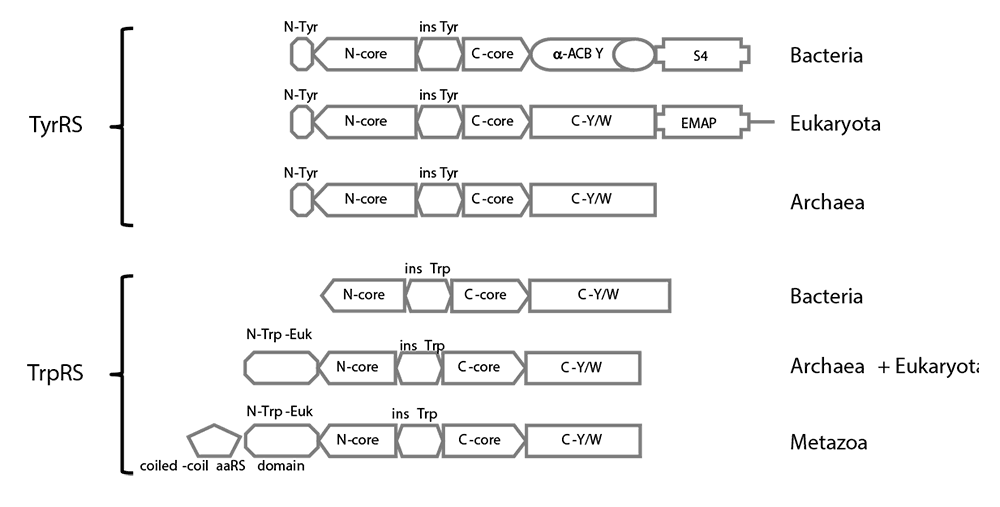
Рис. 3-5. Разнообразие мультидоменной архитектуры гомологичных белков. На схеме сравнивается доменная архитектура двух паралогичных наборов древних и функционально незаменимых для всех организмов ортологичных белков: тирозил-тРНК синтетазы (TyrRS) и триптофанил-тРНК синтетазы (TrpRS). Каждый домен обозначен своей собственной геометрической формой (по Wolf et al., 1999a).
Мультидоменные белки обнаружены у всех форм жизни, но особенно характерны для сложных многоклеточных эукариот (Koonin et al., 2000a; Koonin et al., 2000b). Доменная архитектура этих белков демонстрирует различную степень эволюционной пластичности. Изменчивость особенно выражена у белковых архитектур, включающих так называемые «неразборчивые домены» (promiscuous domains), имеющие склонность к слиянию с разнообразными другими доменами (Basu et al., 2009). Разнообразная мультидоменная архитектура белков запутывает понятие ортологии. Считается, что в ходе долгой эволюции ортологичные гены сохраняют свою уникальность, в том числе функциональную (имеют одну и ту же эволюционную историю). Однако это правило нарушается в тех случаях, когда гены, казалось бы подпадающие под определение ортологии (см. табл. 3-1), меняют доменную архитектуру (см. рис. 3-5): в этих случаях лишь части соответствующих белков в разных организмах имеют одну и ту же эволюционную историю и выполняют одни и те же функции (хотя второе и не может быть гарантировано, поскольку взаимодействие доменов вполне может иметь существенные функциональные последствия).
Контраст между эволюционной пластичностью генома и стабильностью индивидуальных генов
Мы видели, что большинство генов в каждом геноме весьма консервативно: гомологи этих генов — чаще всего легко определяемые ортологи — обнаружены у организмов, эволюционно далеких друг от друга. Тем не менее эта поразительная эволюционная устойчивость генов — лишь одна сторона медали сравнительной геномики. Другая же, оборотная сторона — это «текучесть» генного набора и архитектуры геномов всех форм жизни. Геномы прокариот особенно подвержены изменчивости. Наглядным примером этого является сравнение различных штаммов классической модели бактерий, лабораторного штамма K12 и нескольких патогенных штаммов кишечной палочки Escherichia coli (Perna et al., 2001). Последовательности ортологичных генов у этих бактерий почти одинаковы, однако некоторые патогенные штаммы имеют на 30 процентов больше генов, чем штамм K12, и генные наборы патогенных штаммов радикально различаются. Неизбежно возникает заключение, что «лишние» гены, формирующие так называемые островки патогенности, одними штаммами были приобретены, а другими утеряны (в гл. 5 мы еще вернемся к этой теме).
В более общем плане можно измерить дистанцию между геномами, сравнив, с одной стороны, последовательности консервативных генов-маркеров, таких как рРНК или рибосомных (р) белков, а с другой стороны, исследовав ту часть генов, что формирует легко узнаваемые пары совпадающих ортологов (см. табл. 3-1). В отличие от постепенного, относительно медленного изменения нуклеотидной последовательности генов, наблюдается резкое несовпадение генных наборов (см. рис. 3-6). Заметим, что нет никакого противоречия между этим наблюдением и выводом о том, что для значительного большинства генов в геноме бактерии или археи имеются ортологи в некоторых эволюционно удаленных от них организмах. Здесь слово некоторых ключевое, поскольку у многих генов в любом геноме разное эволюционное происхождение и разная история, и потому их ближайшие родственники могут быть обнаружены в разных таксонах (см. гл. 5). Дистанцию между геномами, определяемую как доля общих (ортологичных) генов, можно использовать для описания «геномной вселенной», рассматриваемой далее в этой главе, а также для построения особого рода дерева эволюции (см. гл. 5).
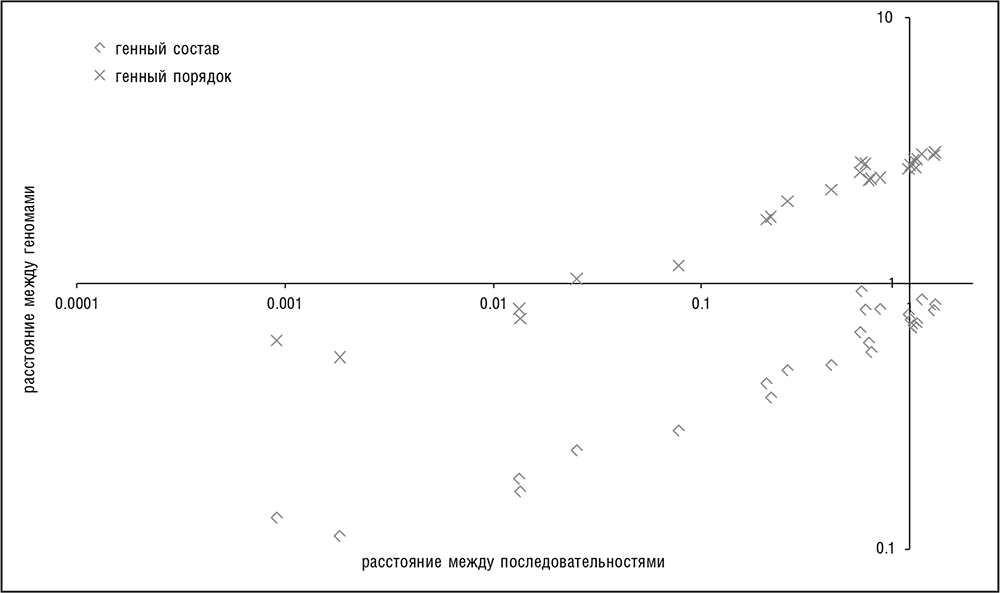
Рис. 3-6. Расхождение порядка генов и генных составов между бактериями по сравнению с расхождением высококонсервативных последовательностей белков. Были вычислены расстояния от кишечной палочки K12, штамм MG1655, до 24 других разнообразных протеобактерий. Расстояние между последовательностями: расстояние наибольшего подобия (maximum likelihood distance) для соединенных выравниваний рибосомных белков вычислено с использованием программы PROTDIST пакета программного обеспечения филогенетического анализа Phylip (Felsenstein, 1996). Расстояние между генными порядками: — ln (JCOG), где JCOG — коэффициент подобия (коэффициент Жаккара) для набора КОГ в двух геномах. Расстояние между генными составами: — ln (JPAIR), где JPAIR — коэффициент Жаккара для множества неупорядоченных пар соседних КОГ в двух геномах. График исполнен в двойных логарифмических координатах.
Геномная архитектура, то есть расположение генов в геноме, проявляет еще большую эволюционную нестабильность, чем генный состав геномов, что контрастирует с консервативностью генных последовательностей (Koonin, 2009a; Novichkov et al., 2009). За исключением организации малых групп функционально связанных генов в оперонах, порядок генов сравнительно слабо сохраняется даже у близкородственных организмов[33]. У прокариот сохранение порядка генов на большом протяжении генов не просматривается даже в некоторых группах геномов, которые сохраняют почти однозначное соответствие ортологичных генов и в среднем более 99 процентов идентичности последовательностей ортологичных белков (см. рис. 3-6). Эукариоты демонстрируют несколько большую сохранность порядка генов. Тем не менее даже в случае эукариот имеется мало общих элементов архитектуры генома между, например, разными типами в царстве животных и вообще никаких между животными и грибами или животными и растениями.
Геномные ландшафты: распределение эволюционных ограничений по разным классам сайтов в геноме
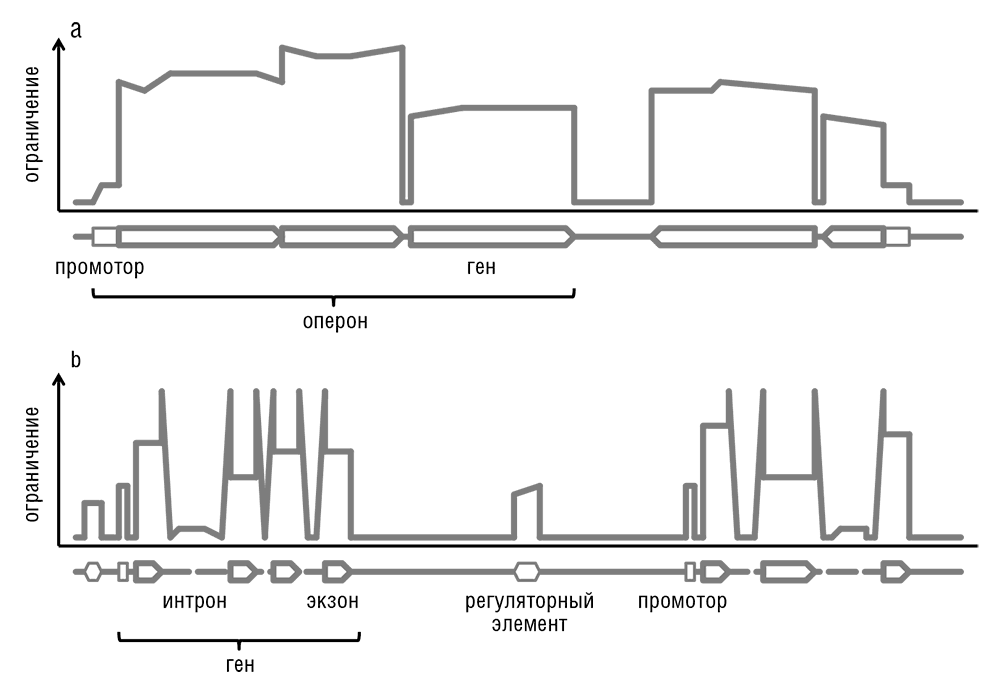
Рис. 3-7. Схематические геномные ландшафты. Распределение эволюционных ограничений по разным сайтам в геномах прокариот и эукариот вскрывает различные принципы геномной архитектуры: а — геном прокариот; б — геном эукариот.
Любой геном может быть представлен в виде геномного ландшафта, графика, напоминающего панораму города, где каждому нуклеотидному сайту присваивается высота, пропорциональная силе влияющих на него эволюционных ограничений. Ограничения имеет смысл рассматривать как меняющиеся в диапазоне от 0 (лишенная ограничений, нейтрально эволюционирущая, функционально не значимая позиция) до 1 (полностью ограниченная, функционально важная позиция, в которой изменения недопустимы, см. рис. 3-7; Koonin and Wolf, 2010b). Распределения ограничений по геному значительно отличаются у форм жизни с различными архитектурами генома. Эти отличия проявляются особенно ярко, если говорить о сравнении, с одной стороны, вирусов и прокариот с их геномами «стена к стене», в основном состоящими из генов, кодирующих белок или РНК, и, с другой стороны, многоклеточных эукариот, в геномах которых кодирующие нуклеотиды находятся в меньшинстве (см. рис. 3-7). В пересчете на один сайт, ограничения в компактных геномах, особенно у прокариот, на несколько порядков сильнее, чем ограничения в больших геномах многоклеточных эукариот. Белок-кодирующие последовательности и последовательности, кодирующие структурные РНК, подвержены наиболее сильным ограничениям во всех геномах. Подавляющее большинство белок-кодирующих генов, особенно у прокариот, имеют низкие значения Ka/Ks, что указывает на сильное давление очищающего отбора на эти последовательности (см. рис. 3-8 и предыдущую главу). В то же время во всех группах организмов существует значительная положительная корреляция между Ka и Ks, указывающая, что даже синонимические сайты в белок-кодирующих генах ограничены примерно в пропорции к ограничениям на несинонимичные сайты (Drummond and Wilke, 2008; см. также гл. 4). Учитывая, что прокариотические геномы почти полностью состоят из белоккодирующих генов со вкраплениями генов структурных РНК и коротких межгенных промежутков, в основном занятых разно образно ограниченными регуляторными регионами, эти компактные геномы содержат мало неограниченных сайтов. Заметным исключением являются псевдогены, редкие у большинства прокариот, но распространенные у некоторых паразитических бактерий, особенно растущих внутри эукариотических клеток, например Rickettsia или Mycobacterium leprae (Harrison and Gerstein, 2002). Геномы большинства вирусов еще более компактны, чем геномы прокариот, причем почти вся последовательность генома занята белок-коди рующими генами.
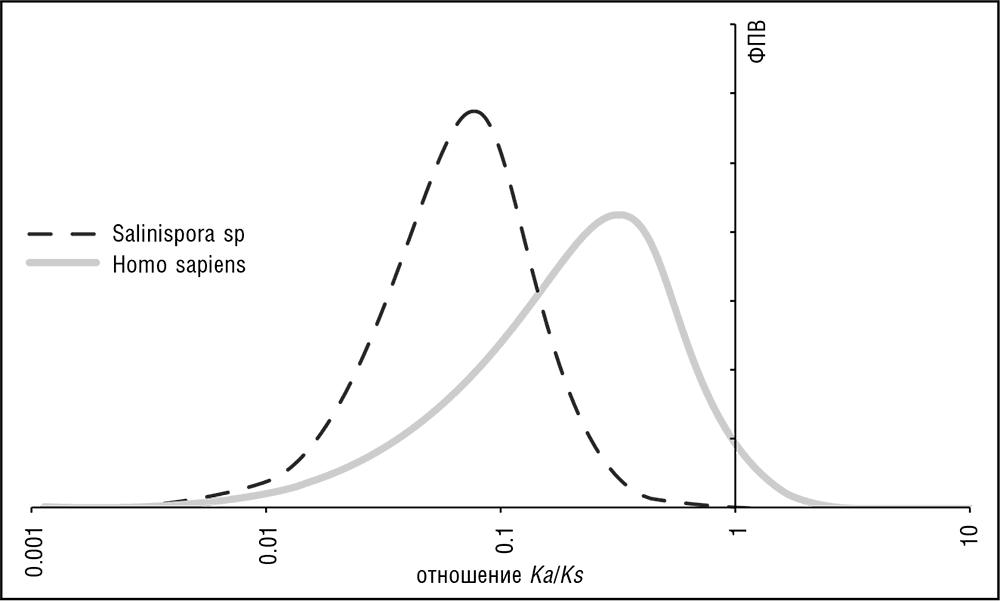
Рис. 3-8. Распределение отношения Ka/Ks в геномах прокариот и эукариот. Salinispora sp.: вычислено по ортологам в S. arenicola CNS-205 и S. tropica CNB-440 (актинобактерии). Homo sapiens: вычислено по ортологам в H. sapiens и Macaca mulatta (приматы). Значения Ка и Ks оценены с использованием программного обеспечения PAML (Yang, 2007). График в логарифмических координатах по оси абсцисс; ФПВ обозначает функцию плотности вероятности.
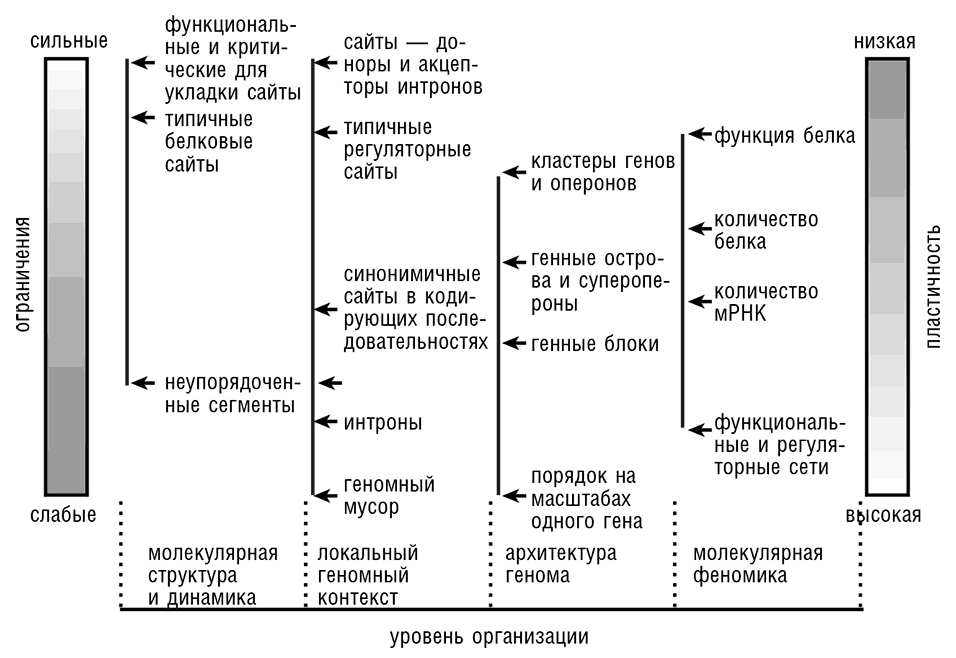
Рис. 3-9. Схематическая сводка эволюционных ограничений, действующих на различные классы геномных сайтов.
Одноклеточные эукариоты, напоминающие прокариот общей архитектурой генома, демонстрируют примерно одинаковые распределения эволюционных ограничений, хотя доля очевидно не подверженных ограничениям некодирующих последовательностей в их геномах несколько выше. Геномы многоклеточных эукариот (растений и особенно животных) являют собою разительную противоположность. Эти организмы имеют богатые интронами геномы с длинными межгенными промежутками; существенная, хотя и переменная часть этих некодирующих последовательностей, по-видимому, эволюционирует, не подвергаясь ограничениям. Доля нуклеотидов в геноме, подверженных эволюционным ограничениям, оценивается методами, основанными на критерии Макдональда—Крейтмана (см. табл. 2-2). Полученные оценки существенно отличаются даже между животными: у Drosophila около 70 процентов нуклеотидных сайтов в геноме, в том числе 65 процентов некодирующих участков, по всей видимости, подвержены отбору (в том числе положительному), а у млекопитающих эта доля оказывается в интервале 3–6 процентов (Koonin and Wolf, 2010b). Примечательно, однако, что абсолютное число подверженных отбору сайтов в столь разных по размеру геномах этих животных довольно близко. Напротив, в Arabidopsis, растении с геномом, размером и общей архитектурой сравнимыми с таковыми Drosophila, доля некодирующих подверженных ограничениям участков, по-видимому, существенно ниже.
Резюмируя существующее понимание ограничений, влияющих на различные классы и сайты во всем известном разнообразии геномов (см. рис. 3-9), отметим, что некоторые фундаментальные простые выводы являются бесспорными. В частности, нет никаких сомнений, что несинонимичные сайты в белок-кодирующих последовательностях и последовательности, кодирующие структурные РНК, являются одними из наиболее сильно ограниченных во всех геномах и что характерное распределение ограничений (геномный ландшафт) сильно коррелирует с архитектурой генома (Koonin and Wolf, 2010b). Однако помимо этих основных принципов, и довольно неожиданно, оказывается, что эволюционные режимы сильно различаются даже для некоторых относительно близких таксонов, таких как членистоногие и позвоночные. Чтобы выработать всеобъемлющую картину эволюционных ограничений и давления, формирующих геном, требуется еще множество дополнительных исследований по различным организмам. В последующих главах рассматриваются различные проявления давления отбора, влияющие на разные части генома.
Вселенная генов
Интеграция результатов сравнительной геномики позволяет нам начать строить карту всей «вселенной генов». Глобальная эволюционная устойчивость генов, проявляющаяся прежде всего в сохранении белковых и РНК-последовательностей, стала очевидной в результате самых первых сравнений секвенированных прокариотических и эукариотических геномов: бактерии Haemophilus influenzae и Mycoplasma genitalium, археи Methanocaldococcus jannaschii и эукариотических дрожжей Saccharomyces cerevisiae (Tatusov et al., 1997). Ключевое обобщение сравнительной геномики состоит в том, что гены не просто сохраняются на различных интервалах эволюционного процесса, но и представляют собой дискретные единицы эволюции, а именно ортологичные эволюционные линии (см. табл. 3-1). В сегодняшней коллекции секвенированных геномов найдены ортологи в далеких таксонах для значительного большинства белок-кодирующих генов в каждом геноме. Ярким примером являются недавние результаты секвенирования генома примитивных животных: многочисленные гены Trichoplax и губки связаны ортологичными отношениями с генами млекопитающих и птиц (Putnam et al., 2007; Srivastava et al., 2008; Srivastava et al., 2010). Один из выводов состоит в том, что характерная продолжительность жизни животного гена в этих линиях охватывает по меньшей мере сотни миллионов лет. Многие другие группы животных, такие как насекомые, утеряли многочисленные гены (Koonin et al., 2004), так что судьба одного и того же гена в большинстве случаев отличается в разных линиях, в результате чего мы получаем «пятнистую» филетическую модель. (Как подчеркивается далее в этой главе, множество поистине универсальных генов чрезвычайно мало.) Судьбы конкретных генов в разных линиях зависят как от случайных факторов, так и от различий в давлении отбора (см. гл. 9). Результаты обширного сравнительного анализа геномов растений, грибов и прокариот полностью совместимы с этим выводом. Когда гены в геноме классифицируются по их относительному «возрасту» (то есть филогенетической глубине, на которой обнаруживаются гомологи), наблюдаемое расхождение подобно для удаленных друг от друга организмов, как показано на рис. 3-3 для генетических наборов человека и грибка Aspergillus fumigatus (Wolf et al., 2009), двух организмов, разделенных, по-видимому, миллиардами лет эволюции. Тем не менее распределения генных возрастов поразительно похожи: в каждом случае древних генов, для которых легко обнаруживаются гомологи в далеких таксонах, значительно больше, чем «молодых» генов. Несмотря на частую потерю в отдельных эволюционных линиях, гены характеризуются чрезвычайной долговечностью, и многие из них, возможно, бессмертны[34].
Как обсуждается далее в этой книге (гл. 5 и 7), пути передачи генетической информации у прокариот принципиально отличаются от таковых у эукариот. Тем не менее доли консервативных генов у них примерно равны. В настоящее время эта доля хорошо известна и очень близка у разнообразных бактерий и архей, почти как фундаментальная постоянная: для 70–80 процентов генов ортологи обнаруживаются в далеких организмах (Koonin and Wolf, 2008b; см. рис. 3-4).
Минимальные наборы генов, замещение неортологичных генов (ЗНОГ) и ускользающее незаменимое ядро жизни
Секвенирование геномов симбиотических и паразитических бактерий привело к соблазнительной идее, что их генетический репертуар может быть близок к «наименьшему возможному набору генов», то есть такому, который является необходимым и достаточным для поддержания простой (прокариотической) клетки при самых благоприятных условиях, какие только могут существовать вне других клеток (Fraser et al., 1995; Mushegian and Koonin, 1996b). Последнее условие чрезвычайно важно, поскольку «наименьшим возможным» набор генов будет лишь в отношении к окружающей среде, в которой соответствующий организм существует (или мог бы существовать, в случае «концептуальных» геномов, полученных компьютерными методами). Однако, как только появились первые два полных бактериальных генома, вторым из которых был геном Mycoplasma genitalium[35], лишенной клеточной стенки паразитической бактерии с размером генома всего около 570 генов, возникла очевидная идея, что «истинный» наименьший набор можно естественным образом вывести, сравнивая геномы этих двух существенно различно специализированных бактериальных патогенов (Mushegian and Koonin, 1996b). Точнее, можно было бы ожидать, что ортологичные гены в двух организмах будет представлять собой набор основных биологических функций, которые необходимы для выживания клетки, независимо от уникального образа жизни каждого организма.
Сравнение геномных наборов H. influenzae и M. genitalium дало 240 пар ортологичных генов, охватывающих большую часть очевидно существенных клеточных функций. Тем не менее в этом консервативном наборе несколько важных функций явно отсутствовали. До сих пор мы не говорили о «настоящей биологии», о биологических функциях, ролях генов, но теперь мы должны начать думать биологически. Определение минимального набора основных биологических функций — задача непростая. Соблазнительно, конечно, попытаться «разобрать эволюцию по винтику»: идя от сравнительной геномики, определить минимальный набор основных генов, сохраняемых во всех клеточных формах жизни. Но этот подход упускает возможность, что разные организмы могли прийти к решению одной и той же принципиальной задачи независимыми путями. Мы увидим далее в этой главе, что такая гипотетическая возможность действительно отражает важный аспект биологической реальности. Таким образом, чтобы очертить минимальный набор клеточных функций, нам необходимо обратиться к логике биохимии и клеточной биологии. Знаний в этих областях несомненно достаточно, чтобы составить разумный каталог основных функций. Само собой, это знание несовершенно, поэтому на самом деле вычисление минимального набора генов требует многократного поочередного обращения к биологическому обоснованию и сравнительному геномному анализу. Мы с Аркадием Мушегяном предположили, что принципиально важные функции, отсутствующие среди 240 ортологов H. influenzae и M. genitalium, вероятно, исполняются неродственными или отдаленно родственными белками в этих двух бактериях. Мы привлекли определенные догадки, чтобы увеличить предполагаемый минимальный набор на 16 дополнительных генов M. genitalium (см. рис. 3-10). Этот простое упражнение в получении минимального набора генов соединением сравнительной геномики и биологической логики оказалось достаточно успешным и, по-видимому, определило приближенный функциональный репертуар простейшей бактериальной клетки, способной к самостоятельному росту в наиболее благоприятных условиях. В самом деле, последующие эксперименты с нокаутом генов подтвердили, что большинство из генов, включенных в минимальный набор, необходимы для выживания бактерий и что гены из минимального набора присутствуют в большинстве (хотя и не обязательно во всех) вновь секвенированных бактериальных геномах (Delaye and Moya, 2010; Koonin, 2003).
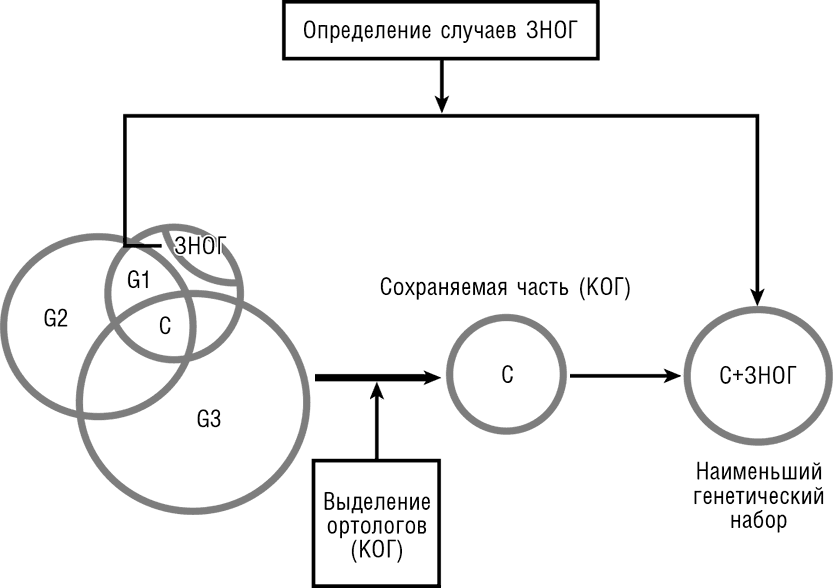
Рис. 3-10. Выделение минимального набора генов клеточной жизни методами сравнительной геномики. G1, G2, G3 — три сравниваемых генома; C — набор консервативных генов.
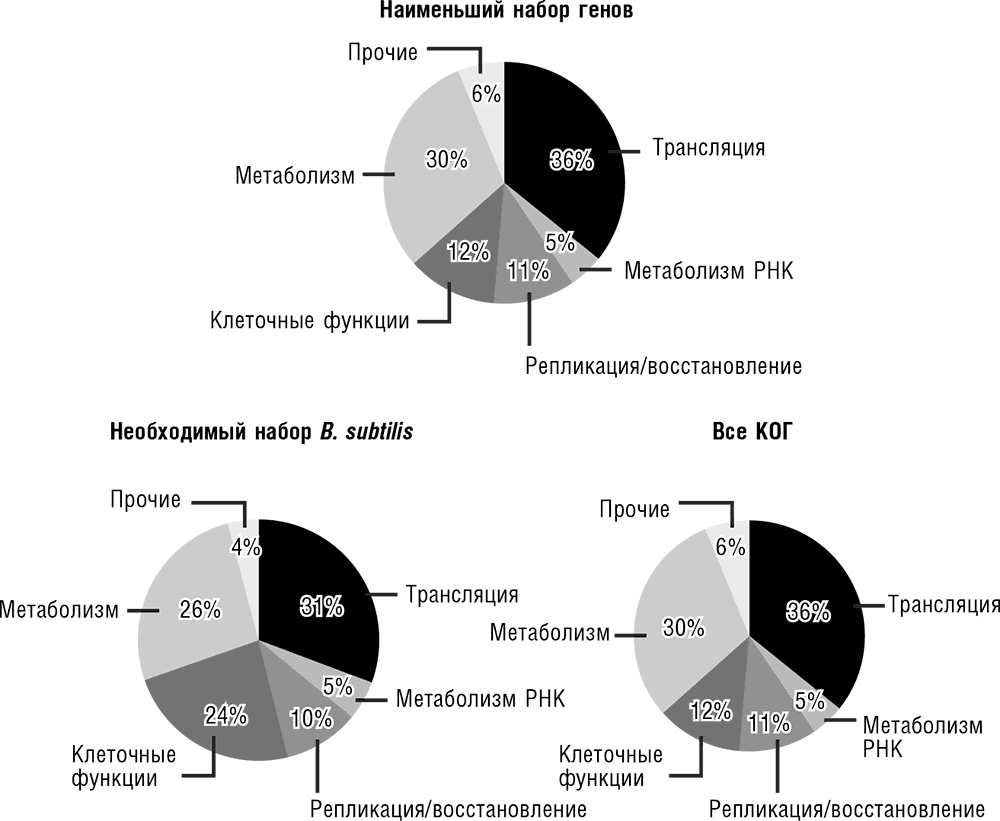
Рис. 3-11. Распределение числа генов по биологическим функциям в минимальном генном наборе, полном наборе КОГ и среди экспериментально определенных незаменимых генов бактерии Bacillus subtilis. Данные по Koonin, 2003.
Поучительно провести теперь функциональную перепись минимального бактериального набора генов. В этом наборе преобладают гены, которые кодируют белки, участвующие в передаче информации в клетке (репликации, транскрипции и, прежде всего, трансляции). Метаболические ферменты и белки транспортной системы представлены куда более разреженно, что вполне ожидаемо для организма, растущего в самой богатой из возможных сред. Этим минимальный набор генов резко отличается от полного набора КОГ, но напоминает набор незаменимых бактериальных генов (нокаут которых убивает бактерию, см. рис. 3-11). Эта особая эволюционная устойчивость систем передачи клеточной информации является одним из центральных обобщений сравнительной геномики. Мы вернемся к этому вопросу позднее.
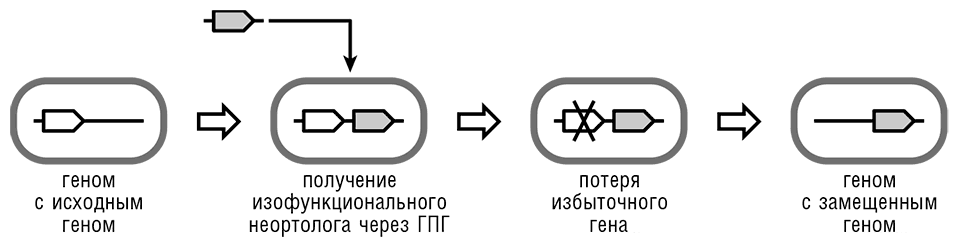
Рис. 3-12. Эволюционный сценарий замещения неортологичных генов.
По-видимому, самым весомым результатом поиска минимального набора генов было открытие, что в списке легко обнаружимых ортологов недостает нескольких важных функций. Этот результат был подтвержден и значительно усилен при сравнении бактериальных геномов с первым архейным (Methanocaldococcus jannaschii), когда в наборе консервативных необходимых функций выявилось несколько дополнительных зияющих пробелов. Эти выводы были обобщены в понятии замещения неортологичных генов (ЗНОГ), эволюционном сценарии, при котором неродственные или отдаленно родственные гены (иными словами, не ортологи) становятся ответственными за одни и те же необходимые функции в разных организмах (Koonin, 2003). Сам этот эволюционный сценарий легко представить (см. рис 3-12): чтобы произошло ЗНОГ, эволюционирующая линия приобретает альтернативный, функционально избыточный ген для некоторой незаменимой функции и таким образом проходит через промежуточное состояние, в котором присутствуют обе реализации данной функции (такая избыточность часто наблюдается у организмов с более сложными геномами), а затем теряет начальный ген (Koonin and Mushegian, 1996). С ростом коллекции секвенированных геномов обнаруживается все больше организмов, в которых оба варианта действительно представлены для разнообразных функций; таким образом, сценарий эволюции ЗНОГ, представленный на рис. 3-12, становится все более правдоподобным.
В табл. 3-2 описывается несколько примеров ключевых биологических функций, для которых два или несколько неродственных ферментов по-разному представлены в частично дополнительных, но обычно перекрывающихся группах эволюционных линий. Даже эти отдельные примеры показывают, что ЗНОГ происходит в самых различных функциональных системах и путях. В дальнейшем, с заметным увеличением числа секвенированных геномов, стало ясно, что ЗНОГ и утрата генов в отдельных линиях настолько широко распространены, что лишь малое число функций являются действительно мономорфными и вездесущими (то есть представлены ортологичными генами во всех организмах). Вместе с тем универсальное ядро жизни уменьшилось почти до исчезновения: все, что остается универсальным, — это около тридцати генов белков трансляции и три больших субъединицы РНК-полимеразы, а также примерно равное число генов структурных РНК (рРНК и тРНК).
Таблица 3-2
Примеры замещения неортологичных генов
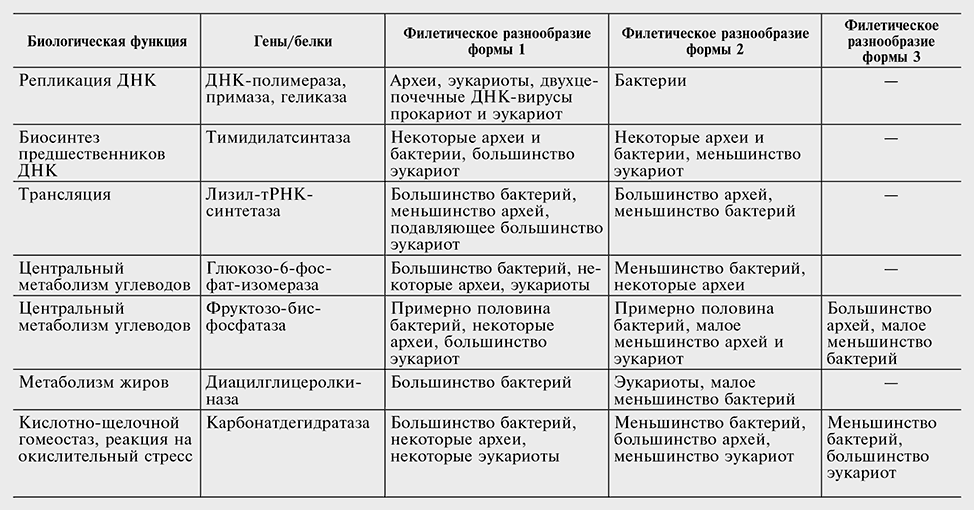
Данные в основном по Omelchenko et al. 2010.
Даже при исключении паразитических бактерий перечень универсальных генов расширяется незначительно (Koonin, 2003). Таким образом, за исключением небольшого числа генов, участвующих в основных этапах передачи информации, не существует универсального генетического ядра жизни, в связи с повсеместными ЗНОГ и потерей генов. Концепция небольшого, универсального набора функций, необходимых для поддержания клетки, остается жизнеспособной, но, учитывая комбинаторику ЗНОГ, этот наименьший набор функциональных ниш может заполняться огромным разнообразием генных ансамблей.
Единицы эволюции и фрактальная структура генетической вселенной
Результаты сравнительной геномики приводят к ключевому обобщению, которое позволяет нам выполнять продуктивные эволюционные исследования: основные единицы эволюции могут быть довольно четко определены, и единицы эти — кластеры ортологичных генов, или эволюционные домены (КОГ), или, еще точнее, линии эволюционирующих ортологичных генов (доменов). Истории отдельных генов часто сложны (а во многих случаях даже чрезвычайно сложны) и включают в себя множественные утраты генов, дупликацию и горизонтальный перенос (ниже в настоящей книге мы обсудим эти явления подробнее, см. гл. 5 и 7). Предрасположенность генов к дупликации, утрате и переносу варьирует в широких пределах. Однако, невзирая на все эти осложнения, атомарное свойство наборов ортологичных генов твердо соблюдается: КОГ суть естественные элементы генетической вселенной.
Генетическая (геномная) вселенная (это только метафора, но удобная и, возможно, продуктивная) может быть представлена как развивающееся пространство-время, заполненное кластерами, состоящими из генов (то есть КОГ), или, точнее, эволюционирующими линиями ортологов, элементарными единицами эволюции. Ортология легче всего прослеживается между прокариотическими генами, так что здесь мы обсудим прокариотическую область геномной вселенной. Тенденции среди эукариотов в принципе похожи, но осложнены распространенной мультидоменной организацией белков и обширной паралогией. В нашем геномном пространстве заметно характерное распределение КОГ по геномам, хорошо аппроксимируемое тремя экспонентами с разными показателями, которые делят генную популяцию на три класса (см. рис. 3-13, а—в; Koonin and Wolf, 2008b).
1. (Почти) универсальные гены, те, что представлены в (почти) всех геномах клеточных форм жизни, составляют лишь малую часть генетической вселенной: это ядро клеточной жизни состоит, самое большее, из приблизительно 70 генов. В каждом конкретном геноме доля этих «ядерных» генов составляет не более 10 процентов, если говорить о самых маленьких геномах клеточных форм жизни (паразитических бактерий, таких как M. genitalium), но обычно ближе к 1 проценту или менее от общего числа генов (см. рис. 3-14).
2. Умеренно консервативная генная оболочка состоит из КОГ, представленных в самых разнообразных геномах, но не в подавляющем их большинстве. Недавний анализ имеющихся прокариотических геномов дает число КОГ оболочки около 5000. Гены оболочки составляют большую часть числа генов в любом геноме (см. рис. 3-14).
3. Малоконсервативное «облако» состоит из КОГ, встречающихся в узких группах организмов, и «генов-сирот» — генов в открытых рамках считывания (ОРС), обнаруженных пока что в одном-единственном геноме, но гомологи которых обычно обнаруживаются во вновь появляющихся геномных данных. Гены «облака» составляют переменную долю в каждом геноме, обычно в интервале 10–30 процентов от общего числа генов (см. рис. 3-14).
Примечательно, что эта структура является самоподобной, или фрактальной: те же три компонента — крошечное ядро, сравнительно большая оболочка и огромное «облако» — проявляются на любом уровне, где бы ни рассекалось генное пространство-время, от всего мира прокариот и до узких групп бактерий (см. рис. 3-14). Мы возвратимся к последствиям этой фрактальности геномного пространства-времени прокариот в главе 5. Заметим, однако, что эволюционная модель, которая объясняла бы наблюдаемую фрактальность, еще ожидает своей разработки[36].
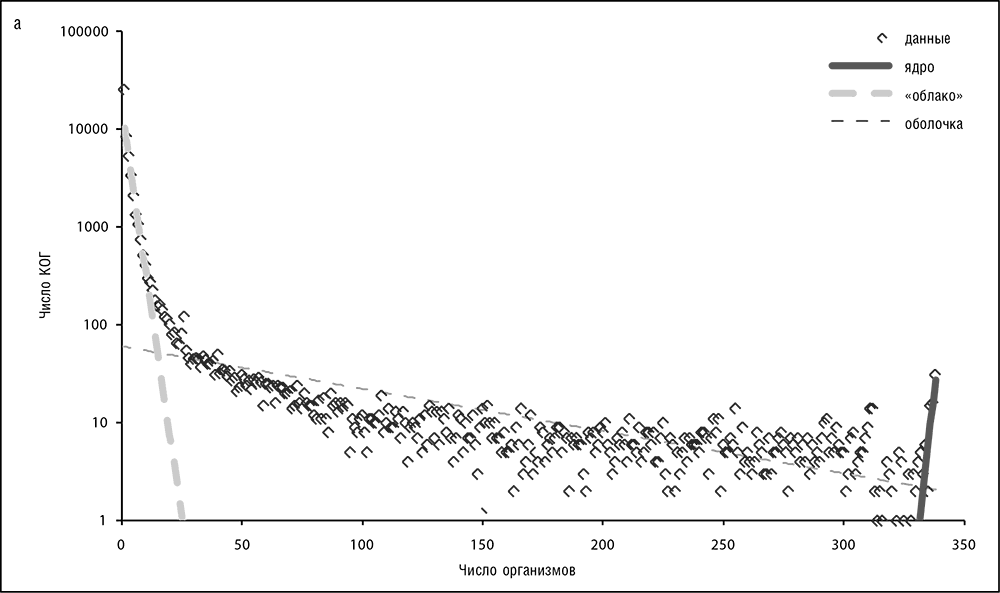
Рис. 3-13 а. Глубокий уровень: 338 прокариот из базы данных EggNOG (Jensen et al., 2008)
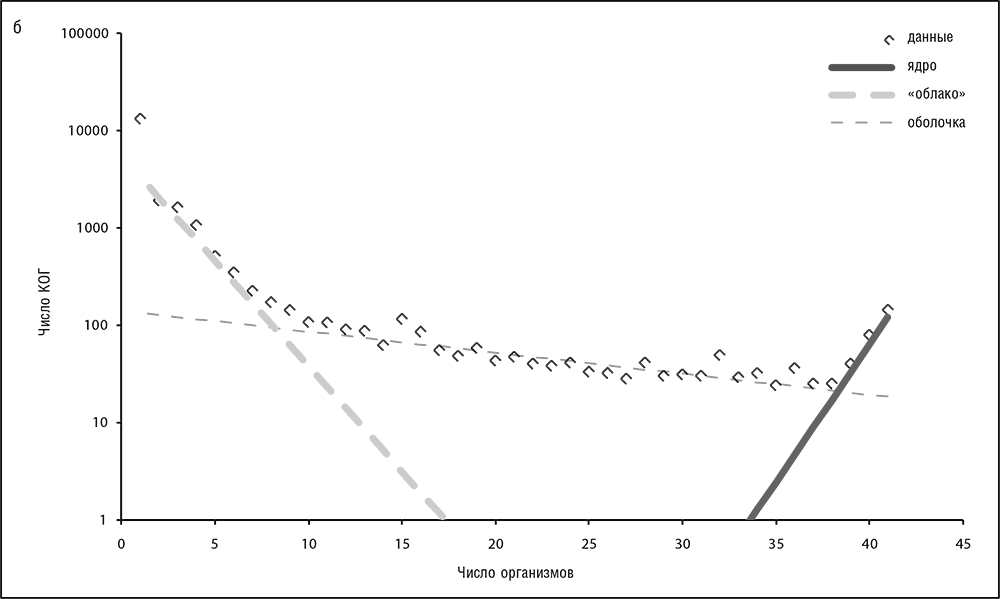
Рис. 3-13 б. Средний уровень: 41 архей из базы данных arCOG (Makarova et al., 2007b)
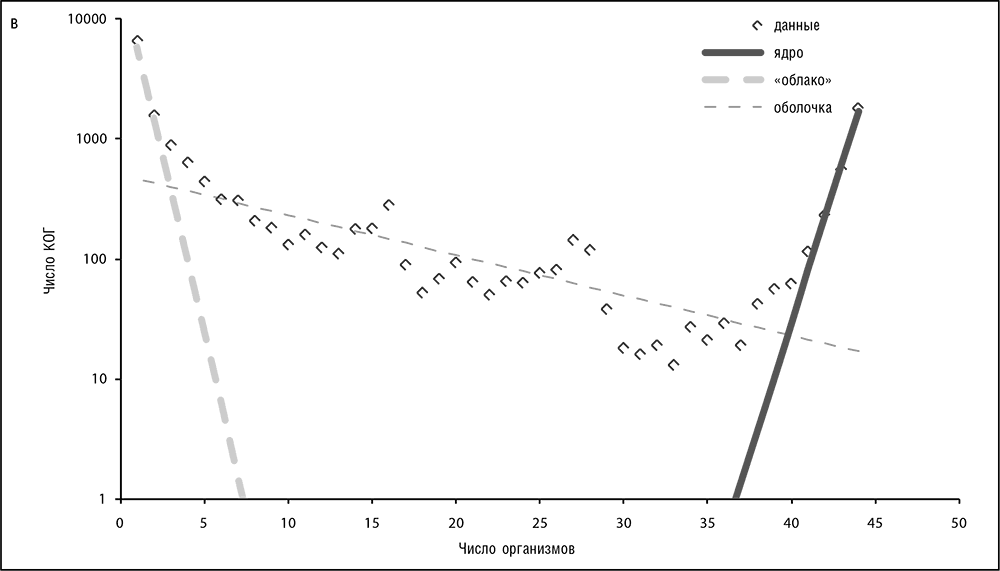
Рис. 3-13 в. Мелкий уровень: 44 вида Escherichia, Shigella и Salmonella из базы данных COG (Tatusov et al., 2003). Регрессия данных экспоненциальными функциями на всех трех рисунках (Koonin and Wolf, 2008b) изображена пунктирными и непрерывными линиями.

Рис. 3-14. Вклад ядра, оболочки и «облака» в состав индивидуальных геномов и генной вселенной как целого. Расчет произведен по данным из базы EggNOG (Jensen et al., 2008). A. fulgidus — архея Archaeoglobus fulgidus; B. subtilis — бактерия Bacillus subtilis.
Более 800 000 книг и аудиокниг! 📚
Получи 2 месяца Литрес Подписки в подарок и наслаждайся неограниченным чтением
ПОЛУЧИТЬ ПОДАРОКДанный текст является ознакомительным фрагментом.
Читайте также
Сравнительная редкость цветов
Сравнительная редкость цветов Среди обитателей умеренного пояса сильно распространено мнение, что пышная растительность тропиков должна порождать также и великолепную флору цветов; такой взгляд подтверждается и многочисленностью крупных красивых цветов, разводимых
ОТ СТРУКТУРЫ К ФУНКЦИИ (функциональная геномика)
ОТ СТРУКТУРЫ К ФУНКЦИИ (функциональная геномика) Недостаточно знания, необходимо также применение; недостаточно хотеть, надо и делать. И. Гёте Хорошо известно выражение, что от слова «халва» во рту слаще не становится. Так же обстоит дело и с нашим геномом. В нем есть
ОТ ГЕНА И БЕЛКА — К ЛЕЧЕНИЮ (медицинская геномика)
ОТ ГЕНА И БЕЛКА — К ЛЕЧЕНИЮ (медицинская геномика) Медицина поистине есть самое благородное из всех искусств. Гиппократ Самые худшие болезни — не смертельные, а неизлечимые. Эбнер-Эшенбах Известный русский биолог Н. Тимофеев-Ресовский писал: «В удивительном по своей
ВСЕ ПОЗНАЕТСЯ В СРАВНЕНИИ (сравнительная геномика)
ВСЕ ПОЗНАЕТСЯ В СРАВНЕНИИ (сравнительная геномика) Между двумя представителями рода человеческого сходства меньше, чем между двумя различными животными. Мишель де Монтень То, что ново в себе, будет понято только по аналогии со старым. Ф. Бэкон Как уже говорилось,
Генетические ландшафты (геногеография)
Генетические ландшафты (геногеография) Анализ нуклеотидных последовательностей геномов позволяет разными методами оценивать степень родства между разными популяциями ныне живущих людей. Осуществляют это, например, по специальным картам, на которые нанесена
ОТ СТРУКТУРЫ К ФУНКЦИИ (функциональная геномика)
ОТ СТРУКТУРЫ К ФУНКЦИИ (функциональная геномика) Недостаточно знания, необходимо также применение; недостаточно хотеть, надо и делать. И. Гёте Хорошо известно выражение, что от слова «халва» во рту слаще не становится. Так же обстоит дело и с нашим геномом. В нем есть много
ОТ ГЕНА И БЕЛКА — К ЛЕЧЕНИЮ (медицинская геномика)
ОТ ГЕНА И БЕЛКА — К ЛЕЧЕНИЮ (медицинская геномика) Медицина поистине есть самое благородное из всех искусств. Гиппократ Самые худшие болезни — не смертельные, а неизлечимые. Эбнер-Эшенбах Известный русский биолог Н. Тимофеев-Ресовский писал: «В удивительном по своей
ВСЕ ПОЗНАЕТСЯ В СРАВНЕНИИ (сравнительная геномика)
ВСЕ ПОЗНАЕТСЯ В СРАВНЕНИИ (сравнительная геномика) Между двумя представителями рода человеческого сходства меньше, чем между двумя различными животными. Мишель де Монтень То, что ново в себе, будет понято только по аналогии со старым. Ф. Бэкон Как уже говорилось,
Генетические ландшафты (геногеография)
Генетические ландшафты (геногеография) Анализ нуклеотидных последовательностей геномов позволяет разными методами оценивать степень родства между разными популяциями ныне живущих людей. Осуществляют это, например, по специальным картам, на которые нанесена
Сравнительная геномика и ранняя эволюция животных[61]
Сравнительная геномика и ранняя эволюция животных[61] Если не учитывать редкую и плохо изученную мелочь вроде трихоплакса, то самыми примитивными животными можно назвать губок, у которых еще нет настоящих тканей, нервной системы и кишечника. Губки противопоставляются
Глава 4 Геномика, системная биология и универсалии эволюции: эволюция генома как феномен статистической физики
Глава 4 Геномика, системная биология и универсалии эволюции: эволюция генома как феномен статистической физики Пер. К. МуратовойВ предыдущей главе была подчеркнута относительная стабильность отдельных генов, составляющая яркий контраст динамизму геномной эволюции.
Глава 5 Сетевая геномика мира прокариот: вертикальные и горизонтальные потоки генов, мобиломы и динамика пангеномов
Глава 5 Сетевая геномика мира прокариот: вертикальные и горизонтальные потоки генов, мобиломы и динамика пангеномов Пер. В. АнисимоваКогда Дарвин писал об эволюции, он имел в виду животных и растения, по крайней мере он использовал эти сложные многоклеточные организмы во
Глава 19 СРАВНИТЕЛЬНАЯ ПСИХОФИЗИОЛОГИЯ
Глава 19 СРАВНИТЕЛЬНАЯ ПСИХОФИЗИОЛОГИЯ Сравнительная психофизиология – наука, нацеленная на установление закономерностей и выявление различий в структурной и функциональной организации мозга, поведении и психике у животных разных видов, в том числе и у человека.
СРАВНИТЕЛЬНАЯ АНАТОМИЯ
СРАВНИТЕЛЬНАЯ АНАТОМИЯ Биологам, занимающимся сравнительной анатомией, давно известно, что виды, образующие более крупные группы (тип, класс или отряд), имеют в своем строении много схожих черт. Развитие техники генетического анализа позволило установить более прочные
СРАВНИТЕЛЬНАЯ СИМВОЛИКА ГЕНОВ
СРАВНИТЕЛЬНАЯ СИМВОЛИКА ГЕНОВ Читатели, которые интересуются литературой по генетике, рано или поздно сталкиваются с проблемой путаницы в обозначениях генов. Дело в том, что различные авторы пользуются различными символами для обозначения одного и того же гена. Это