Глава 22. Метаболизм холестерола. Биохимия атеросклероза
Глава 22. Метаболизм холестерола. Биохимия атеросклероза
Холестерол – стероид, характерный только для животных организмов. Основное место его образования в организме человека – печень, где синтезируется 50% холестерола, в тонком кишечнике его образуется 15–20%, остальное количество синтезируется в коже, коре надпочечников и половых железах. Источники формирования фонда холестерола и пути его расходования представлены на рис 22.1.

Рис. 22.1. Формирование и распределение фонда холестерола в организме.
Холестерол организма человека (суммарное количество около 140 г) условно можно разделить на три пула:
1. пул А (~ 30 г), быстрообменивающийся, состоит из ХС кишечной стенки, плазмы крови, печени и других паренхиматозных органов, обновление происходит за 30 сут (1 г/сут);
2. пул Б (~ 50 г), медленнообменивающийся ХС остальных органов и тканей;
3. пул В (~ 60 г), очень медленнообменивающийся ХС спинного и головного мозга, соединительной ткани, скорость обновления исчисляется годами.
Синтез холестерола происходит в цитозоле клеток. Это один из самых длинных метаболических путей в организме человека. Он проходит в 3 этапа: первый заканчивается образованием мевалоновой кислоты, второй – образованием сквалена (углеводород линейной структуры, состоящий из 30 углеродных атомов). В ходе третьего этапа сквален превращается в молекулу ланостерола, далее происходит 20 последовательных реакций, превращающих ланостерол в холестерол.
В некоторых тканях гидроксильная группа холестерола этерифицируется с образованием эфиров. Реакция катализируется внутриклеточным ферментом АХАТ (ацилКоА:холестеролацилтрансферазой). Реакция этерификации происходит также в крови в ЛПВП, где находится фермент ЛХАТ (лецитин:холестеролацилтрансфераза). Эфиры холестерола – форма, в которой он транспортируется кровью или депонируется в клетках. В крови около 75% ХС находится в виде эфиров.
Регуляция синтеза холестерола осуществляется путем влияния на активность и количество ключевого фермента процесса – 3-гидрокси-3-метилглутарил-КоА-редуктазы (ГМГ-КоА-редуктазы).
Это достигается двумя способами:
1. Фосфорилирование/дефосфорилирование ГМГ-КоА-редуктазы. Инсулин стимулирует дефосфорилирование ГМГ-КоА-редуктазы, переводя её тем самым в активное состояние. Следовательно, в абсорбтивный период синтез ХС увеличивается. В этот период увеличивается и доступность исходного субстрата для синтеза – ацетил-КоА. Глюкагон оказывает противоположное действие: через протеинкиназу А стимулирует фосфорилирование ГМГ-КоА-редуктазы, переводя её в неактивное состояние. В результате синтез ХС в постабсорбтивном периоде и при голодании ингибируется.
2. Ингибирование синтеза ГМГ-КоА-редуктазы. ХС (конечный продукт метаболического пути) снижает скорость транскрипции гена ГМГ-КоА-редуктазы, подавляя таким образом собственный синтез, аналогичный эффект вызывают и жёлчные кислоты.
Транспорт холестерола кровью осуществляется в составе ЛП. ЛП обеспечивают поступление в ткани экзогенного ХС, определяют его потоки между органами и выведение из организма. Экзогенный ХС доставляется в печень в составе остаточных ХМ. Там вместе с синтезированным эндогенным ХС он формирует общий фонд. В гепатоцитах ТАГ и ХС упаковываются в ЛПОНП, и в таком виде секретируются в кровь. В крови ЛПОНП под действием ЛП-липазы, гидролизующей ТАГ до глицерола и жирных кислот, превращаютя сначала в ЛППП, а затем и в ЛПНП, содержащие до 55% ХС и его эфиров. ЛПНП – основная транспортная форма ХС, в которой он доставляется в ткани (70% ХС и его эфиров в крови находится в составе ЛПНП). Из крови ЛПНП поступают в печень (до 75%) и другие ткани, которые имеют на своей поверхности рецепторы ЛПНП.
Если количество ХС, поступающего в клетку, превышает её потребность, то синтез рецепторов ЛПНП подавляется, что уменьшает поток ХС из крови. При снижении концентрации свободного ХС в клетке, наоборот, синтез рецепторов активируется. В регуляции синтеза рецепторов ЛПНП участвуют гормоны: инсулин, трийодтиронин и половые гормоны увеличивают образование рецепторов, а глюкокортикоиды – уменьшают.
В так называемом «обратном транспорте холестерола», т.е. пути, обеспечивающем возвращение ХС в печень, основную роль играют ЛПВП. Они синтезируются в печени в виде незрелых предшественников, которые практически не содержат ХС и ТАГ. В крови предшественники ЛПВП насыщаются ХС, получая его из других ЛП и мембран клеток. В переносе ХС в ЛПВП участвует фермент ЛХАТ, находящийся на их поверхности. Этот фермент присоединяет остаток жирной кислоты от фосфатидилхолина (лецитина) к ХС. В результате образуется гидрофобная молекула эфира холестерола, которая перемещается внутрь ЛПВП. Таким образом, незрезые ЛПВП, обогащаясь ХС, превращаются в ЛПВП3 – зрелые и более крупные по размерам частицы. ЛПВП3 обменивают эфиры холестерола на ТАГ, содержащиеся в ЛПОНП и ЛППП при участии специфического белка, переносящего эфиры холестерола между липопротеинами. При этом ЛПВП3 превращаются в ЛПВП2, размер которых увеличивается за счет накопления ТАГ. ЛПОНП и ЛППП под действием ЛП-липазы превращаются в ЛПНП, которые в основном и доставляют ХС в печень. Небольшая часть ХС доставляется в печень ЛПВП2 и ЛППП.
Синтез жёлчных кислот. В печени из ХС синтезируется 500–700 мг жёлчных кислот в сутки. Их образование включает реакции введения гидроксильных групп при участии гидроксилаз и реакции частичного окисления боковой цепи ХС (Рис. 22.2):
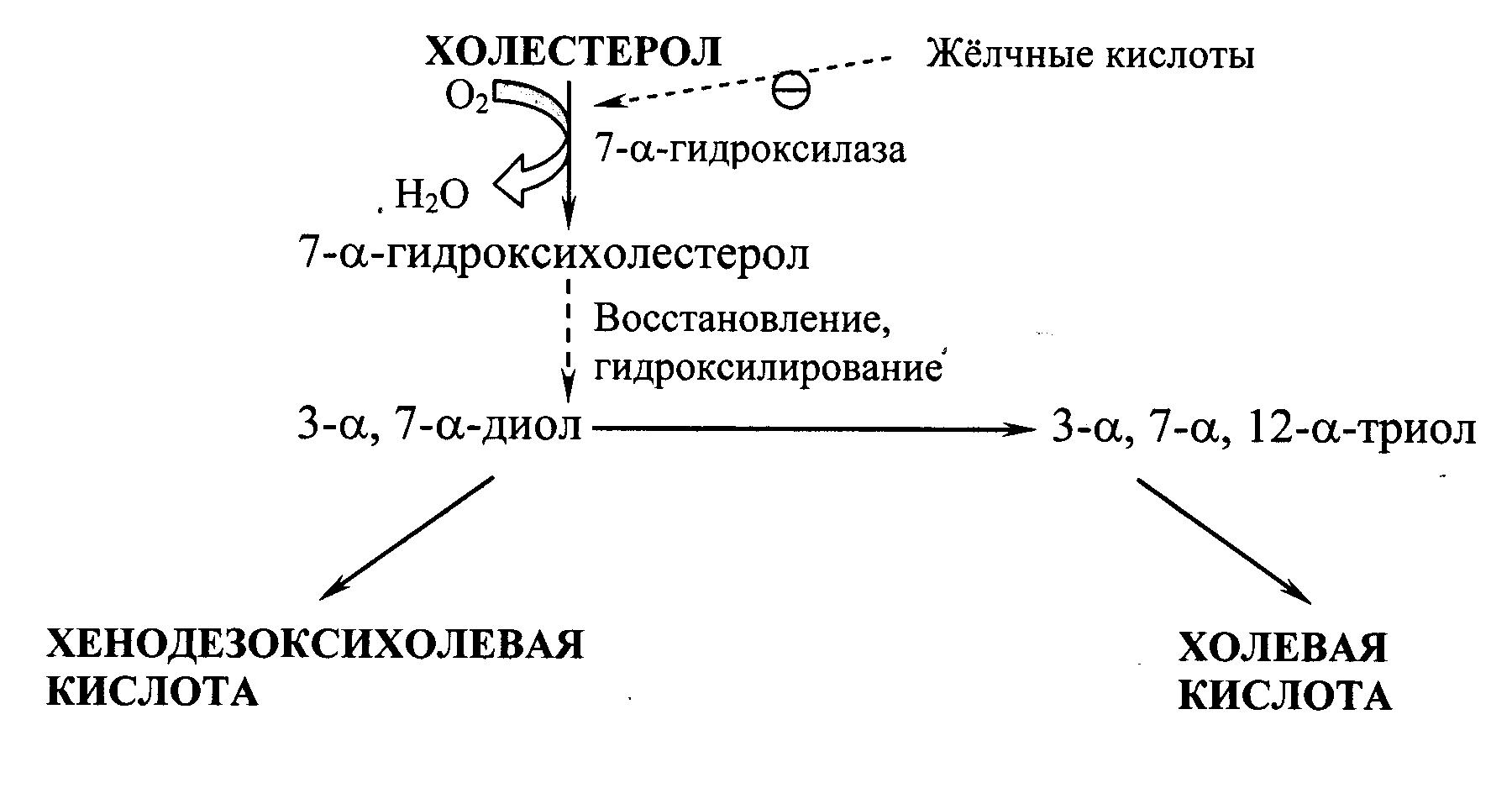
Рис. 22.2. Схема образования жёлчных кислот.
Первая реакция синтеза – образование 7-a-гидроксихолестерола – является регуляторной. Активность фермента, катализирующего эту реакцию, ингибируется конечным продуктом пути – жёлчными кислотами. Еще одним механизмом регуляции является фосфорилирование/дефосфорилирование фермента (активна фосфорилированная форма 7-a-гидроксилазы). Возможна и регуляция путем изменения количества фермента: ХС индуцирует транскрипцию гена 7-a-гидроксилазы, а жёлчные кислоты репрессируют. Тиреоидные гормоны индуцируют синтез 7-a-гидроксилазы, а эстрогены – репрессируют. Такое влияние эстрогенов на синтез жёлчных кислот объясняет, почему желчнокаменная болезнь встречается у женщин в 3–4 раза чаще, чем у мужчин.
Образовавшиеся из ХС холевую и хенодезоксихолевую кислоты называют «первичными жёлчными кислотами». Основная масса этих кислот подвергается коньюгации – присоединению молекул глицина или таурина к карбоксильной группе жёлчной кислоты. Коньюгация начинается с образования активной формы желчных кислот – производных КоА, затем присоединяются таурин или глицин, и в результате образуется 4 варианта коньюгатов: таурохолевая и таурохенодезоксихолевая, гликохолевая и гликохенодезоксихолевая кислоты. Они являются значительно более сильными эмульгаторами, чем исходные жёлчные кислоты. Коньюгатов с глицином образуется в 3 раза больше, чем с таурином, так как количество таурина в организме ограничено. В кишечнике небольшое количество коньюгатов первичных жёлчных кислот под действием ферментов бактерий превращаются во вторичные жёлчные кислоты. Дезоксихолевая кислота, образующаяся из холевой, и литохолевая, образующаяся из дезоксихолевой, хуже растворимы и медленнее всасываются в кишечнике.
Около 95% жёлчных кислот, попавших в кишечник, возвращаются в печень через воротную вену, затем опять секретируются в жёлчь и повторно используются в эмульгировании жиров. Этот путь жёлчных кислот называется энтерогепатической циркуляцией. С фекалиями в основном удаляются вторичные жёлчные кислоты.
Желчнокаменная болезнь (ЖКБ) – патологический процесс, при котором в жёлчном пузыре образуются камни, основу которых составляет ХС.
Выделение ХС в жёлчь должно сопровождаться пропорциональным выделением жёлчных кислот и фосфолипидов, удерживающих гидрофобные молекулы ХС в мицеллярном состоянии. Причинами, приводящими к изменению соотношения жёлчных кислот и ХС в жёлчи являются: пища, богатая ХС, высококалорийное питание, застой жёлчи в жёлчном пузыре, нарушение энтерогепатической циркуляции, нарушения синтеза жёлчных кислот, инфекции жёлчного пузыря.
У большинства больных ЖКБ синтез ХС увеличен, а синтез жёлчных кислот из него замедлен, что приводит к диспропорции количества ХС и жёлчных кислот, секретируемых в жёлчь. В итоге ХС начинает осаждаться в жёлчном пузыре, образуя вязкий осадок, который постепенно затвердевает. Иногда он пропитывается билирубином, белками и солями кальция. Камни могут состоять только из ХС (холестериновые камни) или из смеси ХС, билирубина, белков и кальция. Холестериновые камни обычно белого цвета, а смешанные – коричневые разных оттенков.
В начальной стадии образования камней можно применять в качестве лекарства хенодезоксихолевую кислоту. Попадая в жёлчный пузырь, она постепенно растворяет холестериновые камни, однако это медленный процесс, длящийся несколько месяцев.
Биохимия атеросклероза
Атеросклероз – это патология, характеризующаяся появлением атерогенных бляшек на внутренней поверхности сосудистой стенки. Одна из основных причин развития такой патологии – нарушение баланса между поступлением холестерола с пищей, его синтезом и выведением из организма. У пациентов, страдающих атеросклерозом, повышены концентрации ЛПНП и ЛПОНП. Существует обратная зависимость между концентрацией ЛПВП и вероятностью развития атеросклероза. Это согласуется с представлениями о функционировании ЛПНП как переносчиков ХС в ткани, а ЛПВП – из тканей.
Базовой метаболической «предпосылкой» развития атеросклероза является гиперхолестеролемия. (повышенное содержание холестерола в крови).
Гиперхолестеролемия развивается:
1. вследствие избыточного поступления ХС, углеводов и жиров;
2. генетической предрасположенности, заключающейся в наследственных дефектах структуры рецепторов ЛПНП или апоВ-100, а также в повышенном синтезе или секреции апоВ-100 (в случае семейной комбинированной гиперлипидемии, при которой в крови повышены концентрации и ХС и ТАГ).
Важную роль в механизмах развития атеросклероза играет модифицирование ЛП. Изменения нормальной структуры липидов и белков в составе ЛПНП делает их чужеродными для организма и поэтому более доступными для захвата фагоцитами.
Модифицирование ЛП может происходить по нескольким механизмам:
1. гликозилирование белков, происходящее при увеличении концентрации глюкозы в крови;
2. перекисная модификация, приводящая к изменениям липидов в липопротеинах и структуры апоВ-100;
3. формирование аутоиммунных комплексов ЛП-антитело (изменённые ЛП могут становиться причиной образования аутоантител).
Модифицированные ЛПНП поглощаются макрофагами. Этот процесс не регулируется количеством поглощенного ХС, как в случае его поступления в клетки через специфические рецепторы, поэтому макрофаги перегружаются ХС и превращаются в «пенистые клетки», которые проникают в субэндотелиальное пространство. Это приводит к формированию липидных пятен или полосок в стенке кровеносных сосудов. На этой стадии эндотелий сосудов может сохранять свою структуру. При увеличении количества пенистых клеток происходит повреждение эндотелия. Повреждение способствует активации тромбоцитов. В результате они секретируют тромбоксан, который стимулирует агрегацию тромбоцитов, а также начинают продуцировать тромбоцитарный фактор роста, стимулирующий пролиферацию гладкомышечных клеток. Последние мигрируют из медиального во внутренний слой артериальной стенки, способствуя таким образом росту бляшки. Далее происходит прорастание бляшки фиброзной тканью, клетки под фиброзной оболочкой некротизируются, а ХС откладывается в межклеточном пространстве. На последних стадиях развития бляшка пропитывается солями кальция и становится очень плотной. В области бляшки часто образуются тромбы, перекрывающие просвет сосуда, что приводит к острому нарушению кровообращения в соответствующем участке ткани и развитию инфаркта.
Более 800 000 книг и аудиокниг! 📚
Получи 2 месяца Литрес Подписки в подарок и наслаждайся неограниченным чтением
ПОЛУЧИТЬ ПОДАРОКДанный текст является ознакомительным фрагментом.
Читайте также
Глава 12 Метаболизм
Глава 12 Метаболизм ХимиотерапияБорьба с бактериальными заболеваниями во многом проще, чем с вирусными. Как уже было показано, бактерии проще размножаются в культуре. Бактерии более уязвимы. Живя вне клетки, они производят ущерб организму, отнимая у него питание либо
Глава 8. Введение в метаболизм
Глава 8. Введение в метаболизм Обмен веществ или метаболизм – это совокупность химических реакций в организме, которые обеспечивают его веществами и энергией, необходимыми для жизнедеятельности. Процесс метаболизма, сопровождающийся образованием более простых
Глава 12. Биохимия гормонов
Глава 12. Биохимия гормонов Гормоны (от греческого hormaino – побуждаю) – это биологически активные вещества, которые выделяются эндокринными клетками в кровь или лимфу и регулируют в клетках-мишенях биохимические и физиологические процессы.В настоящее время предложено
Глава 14. Биохимия питания
Глава 14. Биохимия питания Наука о пище и питании называется нутрициологией (от греч. нутрицио - питание). Нутрициология или наука о питании – это наука о пище, пищевых веществах и других компонентах, содержащихся в продуктах питания, их взаимодействии, роли в поддержании
Метаболизм фруктозы
Метаболизм фруктозы Значительное количество фруктозы, образующее при расщеплении сахарозы, прежде чем поступить в систему воротной вены, превращается в глюкозу уже в клетках кишечника. Другая часть фруктозы всасывается с помощью белка-переносчика, т.е. путем
Метаболизм галактозы
Метаболизм галактозы Галактоза образуется в кишечнике в результате гидролиза лактозы.Нарушение метаболизма галактозы проявляется при наследственном заболевании – галактоземии. Оно является следствием врожденного дефекта фермента
Биохимия атеросклероза
Биохимия атеросклероза Атеросклероз – это патология, характеризующаяся появлением атерогенных бляшек на внутренней поверхности сосудистой стенки. Одна из основных причин развития такой патологии – нарушение баланса между поступлением холестерола с пищей, его
Биохимические основы лечения атеросклероза.
Биохимические основы лечения атеросклероза. Важным лечебным фактором, снижающим риск развития гиперхолестеролемии и атеросклероза, является гипокалорийная и гипохолестериновая диета, Поступление ХС с пищей не должно превышать 300 мг/сут. К лечебным и профилактическим
Глава 25. Метаболизм отдельных аминокислот
Глава 25. Метаболизм отдельных аминокислот Метаболизм метионина Метионин – незаменимая аминокислота. Метильная группа метионина – мобильный одноуглеродный фрагмент, используемый для синтеза ряда соединений. Перенос метильной группы метионина на соответствующий
Глава 28. Биохимия печени
Глава 28. Биохимия печени Печень занимает центральное место в обмене веществ и выполняет многообразные функции:1. Гомеостатическая - регулирует содержание в крови веществ, поступающих в организм с пищей, что обеспечивает постоянство внутренней среды организма.2.
Глава 30. Биохимия крови
Глава 30. Биохимия крови Кровь – жидкая подвижная ткань, перемещающаяся по сосудам. Выполняет роль транспортного и коммуникативного средства, интегрирующего обмен веществ в различных органах и тканях в единую систему. Общая характеристика Общий объем крови у взрослого
Глава 31. Биохимия почек
Глава 31. Биохимия почек Почка – парный орган, основной структурной единицей которого является нефрон. Благодаря хорошему кровоснабжению почки находятся в постоянном взаимодействии с другими тканями и органами и способны влиять на состояние внутренней среды всего
Глава 33. Биохимия мышечной ткани
Глава 33. Биохимия мышечной ткани Подвижность является характерным свойством всех форм жизни - расхождение хромосом в митотическом аппарате клеток, воздушно-винтовые движения жгутиков бактерий, крыльев птиц, точные движения человеческой руки, мощная работа мышц ног. Все
Глава 34. Биохимия соединительной ткани
Глава 34. Биохимия соединительной ткани Соединительная ткань составляет около половины от сухой массы тела. Все разновидности соединительной ткани, несмотря на их морфологические различия, построены по общим принципам:1. Содержит мало клеток в сравнении с другими
Глава 2 Биохимия экономики
Глава 2 Биохимия экономики Также любят они соседа и жмутся к нему, ибо им необходимо тепло. Ницше Ф. Так говорил Заратустра Как правило, люди отвечают добром на добро и испытывают непроизвольную симпатию к тем, кто относится к ним хорошо. Это естественное чувство симпатии
Глава 9. Мембраны и биохимия
Глава 9. Мембраны и биохимия Электронный микроскоп показал, что биохимические реакции в живой клетке протекают с активным участием мембранных процессов. Это заключение относится и к нервной, и к глиальной клетке, и к внутриклеточным органеллам.Следует признать, что