Глава 12. СТИМУЛЯЦИЯ БЕЗ ПРИЛОЖЕНИЯ ЭНЕРГИИ
Глава 12.
СТИМУЛЯЦИЯ БЕЗ ПРИЛОЖЕНИЯ ЭНЕРГИИ
Остановимся на мгновение и подведем промежуточные итоги. Мы начали с поисков различий между живыми существами и неживыми предметами и перешли от них к термодинамике тепловых машин.
Затем мы пришли к выводу, что живые существа тепловыми машинами не являются, но при этом вполне вероятно, что энергию они получают в результате процесса, сходного с горением. От этого повествование закономерно перешло к химической энергии и к тому, что она представляет собой с точки зрения законов термодинамики. Это соображение, в свою очередь, вынудило нас объяснить, почему именно реакции типа задействованных при горении и предполагаемых в участии в процессах жизнедеятельности называются в терминах термодинамики «спонтанными», внешне таковыми не являясь.
Таким образом, мы перешли к предмету обсуждения предыдущей главы — энергии активации, необходимой для того, чтобы «запустить» спонтанную реакцию.
Так чем же теперь это может нам помочь в нашей основной задаче — исследовании природы живой материи? Предположим, что в организме человека происходят такие же реакции, как и при горении. Соответственно они должны обладать энергией активации, которую организм должен предоставить прежде, чем эти реакции начнут спонтанно протекать, снабжая при этом, в свою очередь, организм энергией, необходимой для его функционирования. И кажется, что все описанное в предыдущей главе к живой ткани никоим образом относиться не может.
Я описал два способа, которыми реагенты могут получить энергию активации, — с помощью тепла и с помощью света, но в живом организме явно не используется ни то ни другое. Температура живой ткани практически никогда не поднимается выше 37 °С, чего явно недостаточно для запуска реакций, сходных с горением. То же самое со световым излучением — живая ткань в принципе может быть источником света (примером тому служат светлячки), но это никогда не бывает излучение с высоким содержанием энергии, достаточным для активации химических процессов. Так как же решают живые организмы задачу предоставления энергии активации?
Может быть, перед нами наконец-то пример неподчинения живой материи законам термодинамики? Может быть, вот он наконец-то, тот четкий критерий, что позволит нам провести границу между живой и неживой материей?
Перед тем как перейти к непосредственному ответу на вопрос, давайте посмотрим, существуют ли в мире неживой природы явления, в которых спонтанная реакция запускается без поступления энергии активации, необходимой в большинстве случаев. Ведь если они существуют, то и в живой ткани вполне может происходить нечто подобное.
Первый значительный пример такого рода был обнаружен в связи с производством серной кислоты.
Серная кислота, как и ряд других сильнодействующих кислот, была открыта в Средние века, став одним из очень важных, хотя и недооцененных, продуктов алхимических опытов. Алхимики слишком концентрировались на поиске рецептов производства золота и слишком мало внимания обращали на действительно важные результаты своих исследований.
Серная кислота действует во много раз сильнее, чем самая сильнодействующая кислота из известных древним (уксусная кислота виноградного происхождения). Она вступает в ряд химических реакций, которые более слабые кислоты или не могут повторить вообще, или повторяют гораздо медленнее. Таким образом, сильнодействующие кислоты в целом и серная кислота в частности явили собой очень действенный химический инструмент, как для исследовательского, так и для промышленного применения. Даже сегодня серная кислота является самым используемым в химической промышленности веществом, если, конечно, не считать повсеместно присутствующих воздуха и воды, да еще, быть может, соли. Ежегодно изготавливается около пятнадцати миллионов тонн серной кислоты, и иногда даже считается, что масштаб индустриализации страны можно определить по количеству потребляемой ею серной кислоты.
Понятно, что важное значение приобрела разработка технологий дешевого и массового производства серной кислоты. На заре современности ее производство было сложным и дорогим процессом с ограниченным объемом.
Причиной тому были не трудности с добычей исходного сырья. Им служит сера (S) — вещество известное еще в древности, широко распространенное и веками добываемое в достаточном количестве и сравнительно легко, например, в Сицилии.
Сера хорошо горит, вступая в соединение с кислородом и образовывая при этом удушливый газ, сернистый ангидрид (SO2). Именно этот газ и дает знаменитый «запах серы», а не сама сера, которая запаха как раз не имеет.
Сернистый ангидрид растворим в воде, соединяясь затем с молекулой воды в «сернистую кислоту» — H2SO3.
S + O2 ? SO2,
SO2+H2O ? H2SO3.
Действие сернистой кислоты имеет лишь умеренную силу, и к тому же она не очень стабильна. Это не то же самое, что необходимая нам серная кислота, имеющая в своем составе на один атом кислорода больше — H2SO4.
И вот самое сложное — это именно добиться присоединения этого самого последнего атома. Точнее, сернистый ангидрит может и дальше соединяться с кислородом, образовывая при этом серный ангидрид (SO3), а он уже при растворении в воде и даст вожделенную серную кислоту:
2SO2 + O2 ? 2SO3,
SO3 + Н2О ? H2SO4.
На бумаге все выглядит гладко, но проблема состоит в том, что реакция соединения сернистого ангидрида с кислородом, хоть и приводит к снижению уровня свободной энергии, а значит — является спонтанной, вместе с тем требует столь высокой энергии активации, что путем прямого соединения сернистого ангидрида с кислородом удается получить очень мало серного ангидрида. Поэтому на протяжении всего XVII и начала XVIII века серную кислоту производили лишь по чуть-чуть и использовали крайне редко. На получение двух фунтов серной кислоты уходила неделя тяжкого труда.
Затем, в 40-х годах XVIII века, один из производителей серной кислоты, Джошуа Уорд, вдруг обнаружил, что при сжигании стандартной упаковки серы удается произвести гораздо больше серной кислоты, если примешать к ней селитру (азотнокислый натрий). Выяснилось ли это случайно, или изначально планировалось подмешать в серу селитры, чтобы она быстрее сгорала, на манер пороха (который состоит, как известно, из серы, селитры и порошкового угля), я не знаю. Так или иначе, Уорд запатентовал свою технологию, и стоимость производства серной кислоты резко упала.
Масштабы производства этого химиката соответственно возросли, и сам технологический процесс стали осуществлять уже не в хрупких стеклянных колбах, а в больших свинцовых камерах.
На протяжении полувека производители продолжали подмешивать селитру в серу, так и не понимая, зачем именно они это делают. Никаких азотсодержащих веществ в результате реакции никогда не образовывалось. По окончании процесса селитра так и оставалась в камере нетронутой, так что ее можно было использовать снова и снова. В ходе реакции она ничего не отдавала (по крайней мере, этого не обнаруживалось) и не расходовалась. Так в чем же заключалась ее роль?
В 1806 году два французских химика, Шарль Бернар Дезорм и Николя Клеман, выдвинули предположительное объяснение, принцип которого принят и по сей день. При сгорании серы и селитры образуются два газа, сернистый ангидрид (SO2) и перекись азота (NO2). По предположению ученых, перекись азота отдает затем один из своих атомов кислорода на образование серного ангидрида, а сама превращается в простой оксид азота (NO):
SO2 + NO2 ? SO3 + NO.
Сам оксид азота после этого забирает один атом из атмосферного кислорода и снова становится перекисью азота:
2NO + O2 ? 2NO2
Таким образом, перекись азота выполняет роль посредника, отдавая атом кислорода сернистому ангидриду, забирая на его место другой из воздуха и снова передавая его другой молекуле сернистого ангидрида, и так далее. На всем протяжении этой активной работы перекись азота остается сама собой, превращаясь в конце реакции обратно в селитру, как будто ничего с ней и не происходило.
Со времен Дезорма и Клемана эта картина уточнялась и усложнялась. Сейчас уже известно, что перекись азота и сернистый ангидрид соединяются вместе с водой и кислородом в единый сложный «продукт присоединения», который в итоге распадается, в частности, на серную кислоту и селитру. Однако принцип происходящего остается неизменным — сначала селитра участвует в реакции, а затем восстанавливается в прежнем виде.
С точки зрения энергии при такой схеме удается избежать момента приложения мощной энергии активации, необходимой для непосредственного соединения сернистого ангидрида и кислорода (рис. 20). Да и самого непосредственного соединения при такой схеме тоже не происходит. Вместо этого сернистый ангидрид соединяется с перекисью азота, образовывая с ним «продукт присоединения», в ходе реакции, требующей значительно более низкой энергии активации, а образование серной кислоты и восстановление перекиси азота происходит в ходе другой реакции, тоже требующей невысокой энергии активации.
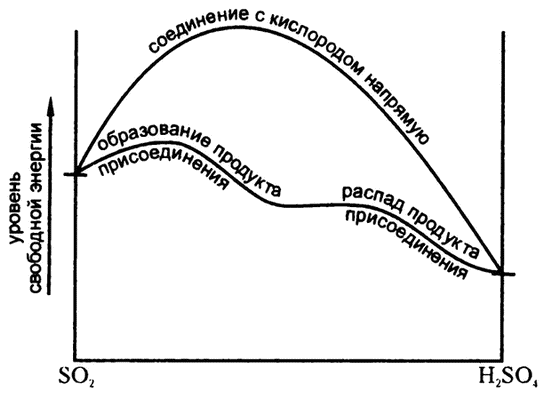
Рис. 20. Как избежать приложения энергии активации
Здесь нет получения чего-либо «ниоткуда». Законы термодинамики не нарушаются. Свободная энергия имеющегося на входе сернистого ангидрида и получаемого на выходе серного ангидрида (или серной кислоты) остается прежней. Общее направление движения реакции на обоих этапах — «вниз», от высокого уровня энергии к низкому. Однако вместо перехода от сернистого ангидрида непосредственно к серному через высокий порог благодаря добавлению селитры путь прокладывается иным образом — через два последовательных низких порога. В терминах энергии активации эта интерпретация впервые прозвучала в отношении всех явлений подобного рода в 1894 году из уст немецкого физика Вильгельма Оствальда.
Если рассматривать реакцию сернистого ангидрида с кислородом для образования серного ангидрида как путь по железной дороге из одной долины в другую, ниже расположенную, через перевал, то добавление селитры равносильно открытию нового пути, через более низкие перевалы. Если поездам хронически не хватает топлива, то через высокий перевал переберутся лишь немногие, и успешное завершение переезда из одной долины в другую станет делом редким и удивительным. Но открытие нового пути позволит облегчить и сделать более массовым движение между долинами даже без поставок дополнительного топлива.
В течение XIX века обнаруживались все новые и новые примеры того, что примешивание определенных веществ в небольших количествах помогало ускорять реакции больших объемов реагентов, при том что сами вещества в процессе реакции не расходуются. В 1812 году русский химик Готлиб Сигизмунд Кирхгоф обнаружил, что если при варке крахмала в воде добавить чуть-чуть разведенной серной кислоты, то крахмал распадается с образованием сахара, полностью идентичного тому, что содержится в винограде (позже его назовут глюкозой).
Если же кислоты не добавлять, а просто варить крахмал в воде — этого не происходит. Более того, при распаде крахмала с помощью кислоты сама кислота не расходуется — окончательный раствор сахара содержит не меньше кислоты, чем исходный раствор крахмала.
А английский химик Гемфри Дэви в 1816 году обнаружил, что наличие проволоки из некоторых металлов стимулирует соединение различных паров, например спиртового, с кислородом. Это оказалось особенно справедливым в отношении редких и инертных металлов, платины и палладия.
Исследования в этом направлении привели к тому, что немецкий химик Иоганн Вольфганг Дёберейнер установил, что участие порошка платины приводит к соединению водорода с кислородом при комнатной температуре. Он изобрел даже автоматическую зажигалку (известную как «лампа Дёберейнера») за много лет до того, как были придуманы спички для практического использования. Основу его «лампы» составляла емкость с кислотой, сконструированная таким образом, чтобы кислоту можно было подавать во внутреннюю камеру, в которой содержалась цинковая полоска. В ходе реакции между цинком и кислотой высвобождался водород и выходил в отверстие в верхней части емкости. Там поток водорода попадал в камеру с платиновым порошком и мгновенно загорался.
Разумеется, платиновый порошок, во-первых, дорог, а во-вторых, быстро теряет свои свойства при подобном использовании из-за наличия посторонних примесей в поступающем водороде. Так что «лампе Дёберейнера» была уготована судьба всего лишь химической игрушки. Однако сам факт ее работы очень наглядно продемонстрировал уникальные свойства платины. Кроме того, стало ясно, что реакции, происходящие при горении (и, возможно, в человеческом организме), можно заставить происходить и при комнатной температуре без обязательного внесения энергии активации (что мы и собирались выяснить в начале главы).
В 1836 году этот факт, а с ним — и ряд других привлекли внимание шведского химика по имени Йене Якоб Берцелиус. В свое время этот человек железной рукой правил миром химии, проявляя интерес (причем с весьма плодотворными результатами!) ко всем разделам этой стремительно развивавшейся науки.
Берцелиус свел воедино все разрозненные факты, которые были ему известны, указал на их значение и предложил дать описываемому явлению название «катализ», от греческого слова, означающего «распад», — в честь первого факта распада крахмала на сахар в присутствии кислоты. Термин был принят и используется до сих пор.
Любое вещество, обладающее свойством, присутствуя в небольших количествах (иногда — в совсем малых), ускорять реакцию между большими количествами других веществ, не расходуясь при этом в ходе реакции, называется катализатор. Кислота — это катализатор реакции распада крахмала на сахар. Платина — это катализатор реакции соединения водорода с кислородом при комнатной температуре. Перекись азота — это катализатор создания серной кислоты.
Кстати, сам процесс производства серной кислоты в свинцовой камере сошел на нет благодаря тем же новым открытиям, благодаря которым и появился. В 1831 году английский химик Перегрин Филлипс обнаружил, что платина является более продуктивным катализатором превращения сернистого ангидрида в серный, чем селитра, и запатентовал свое открытие. К середине XIX века, когда были разработаны методы продления активной жизни дорогого платинового катализатора, новый «контактный процесс», где для соединения с кислородом сернистый ангидрид должен вступить в контакт с платиной, повсеместно вытеснил старую технологию производства кислоты в свинцовой камере.
Сразу хочется назвать свет и тепло также катализаторами, поскольку добавление небольшого количества тепла ускоряет любую реакцию, а добавление небольшого количества света приводит к взрывообразной реакции до того инертной смеси водорода с хлором. На самом деле реакции по типу последней часто так и называют — катализируемыми светом.
Однако термин «катализатор» используют, как правило, только по отношению к веществам.
Понятно, что на самом деле такие катализаторы, как перекись азота или кислота, все же участвуют в реакции, восстанавливаясь впоследствии. Это активные вещества, принимающие полноценное участие в химических реакциях, когда им создают соответствующие условия.
Вот платина — другое дело. Это самый замечательный катализатор из всех открытых в начале XIX века, инертный металл, крайне тяжело вступающий в реакцию с другими веществами лишь под воздействием совсем уж непреодолимых обстоятельств. В чем же тогда заключается его участие во множестве реакций, в результате которых получаются катализированные соединения? Ведь немыслимо же, чтобы платина просто оказывала на молекулы реагирующих веществ какое-то мистическое воздействие.
В 1833 году английский ученый Майкл Фарадей выдвинул на эту тему следующее предположение. Он обнаружил, что кусок чистой платиновой фольги катализирует реакцию соединения водорода с кислородом, но при добавлении к смеси небольшого количества угарного газа (СО) действие катализатора прекращается. Угарный газ как бы «отравляет» платину. (Тут можно увидеть интересную аналогию: стоит подмешать немного угарного газа к нашему собственному запасу кислорода — и он с таким же успехом отравит и нас.)
Фарадей предположил, что кислород с водородом образовывают пленку на поверхности платины. В этой пленке оба вещества гораздо плотнее примыкают друг к другу, чем в исходной смеси газов. В общем, можно сказать, что эффект катализатора в данном случае сходен с эффектом нагнетания давления, ведь известно, что повышение давления в смеси газов зачастую ускоряет их реакцию. Затем Фарадею осталось указать, что угарный газ, даже в малых количествах, тоже склонен образовывать пленку на платине, но поскольку он, в отличие от водорода и кислорода, никуда с нее не девается, то в итоге он полностью закрывает платину и от того и от другого, на чем катализ и заканчивается.
Представление о том, что в некоторых катализируемых реакциях ведущая роль принадлежит поверхности катализатора, до сих пор не опровергнуто, оно лишь периодически уточняется. Так, само по себе наличие газовой пленки на такой поверхности ничего не объясняет, поскольку уже после Фарадея выяснилось, что высокое давление, казалось бы равносильное по механизму действия катализатору, не приводит к столь же резкому ускорению реакции.
В 1916 году американский химик Ирвинг Ленгмюр видоизменил теорию Фарадея и внес в нее предположение о том, что молекулы газовой пленки удерживаются на поверхности настоящими химическими связями с атомами платины.
Крайние атомы платины, находящиеся на поверхности, не со всех сторон окружены другими атомами платины, так что их способность образовывать химические связи задействована не полностью. В сторону открытого пространства атомы платины (Pt) способны образовывать связи с чужими атомами. Соответственно тот факт, что платиновый порошок имеет гораздо более выраженные каталитические свойства, чем просто кусок платины, можно объяснить тем, что на единицу веса порошок имеет гораздо большую площадь поверхности и гораздо больше атомов могут вступать в реакцию с образованием химических связей.
Так что можно рассматривать платину как вещество, все-таки вступающее в полноценную реакцию с кислородом. Все атомы с поверхности платины, вцепляясь в атомы кислорода, образовывают некое подобие «продукта присоединения» — оксид платины. Энергия активации для образования этого оксида крайне мала, как и энергия активации следующей реакции — соединения водорода с кислородом из этого оксида (в отличие от соединения с кислородом из его двухатомных молекул, составляющих газ). Соединение происходит очень быстро, поскольку кинетической энергии молекул газа при комнатной температуре более чем достаточно для того, чтобы предоставить необходимую энергию активации.
После того как соединение произошло, атом платины оказывается соединенным с молекулой воды. Но связь между ним и молекулой достаточно слаба, и бомбардировка молекулами газа легко разрывает ее. Атом платины, таким образом, высвобождается для образования новой связи со следующим атомом кислорода.
«Отравление» поверхности платины угарным газом происходит по причине способности атома платины связываться как с молекулой кислорода, так и с молекулой угарного газа. Но угарный газ, в отличие от кислорода, присоединившись к платине, не вступает в реакцию с водородом (как и ни с чем другим, что бы ни присутствовало в системе) для образования слабо удерживаемой молекулы. Он крепко застревает на своем месте. Каждый атом платины, занятый молекулой угарного газа, так и остается занятым, и так происходит со всеми атомами поверхности, пока платина полностью не утратит свои каталитические свойства.
Даже поверхностный катализ не отменяет и не изменяет основных законов энергообмена в химических реакциях. Он может лишь перенаправить ход реакции таким образом, чтобы она проходила через промежуточные реакции с низким уровнем энергии активации, и ускорить тем самым ход реакции в целом, но катализ не влияет на общие изменения энергии в ходе реакции, и в принципе не может этого сделать.
В частности, это значит, что катализатор не может обратить спонтанную реакцию вспять. Если водород спонтанно соединяется с кислородом, образовывая при этом воду, то катализатор может ускорить этот процесс за счет снижения уровня энергии активации. Но никакой катализатор не может вызывать массовое разложение воды на водород и кислород при комнатной температуре без приложения дополнительной энергии.
Далее, в любой обратимой реакции положение точки равновесия (как вы помните — это точка, в которой уровень свободной энергии минимален) никак не смещается по причине присутствия катализатора. Катализатор может лишь ускорять реакцию в обоих направлениях и способствовать скорейшему достижению точки равновесия. Однако изменить значение самой точки равновесия катализатор не способен ни на йоту.
Теперь, разобравшись с катализаторами и обретя необходимый багаж базовых знаний, мы можем перейти наконец к живой материи. Во второй части книги я постараюсь показать, насколько принципы справедливые и незыблемые для неодушевленного мира применимы к процессам, происходящим в человеческом организме.
Более 800 000 книг и аудиокниг! 📚
Получи 2 месяца Литрес Подписки в подарок и наслаждайся неограниченным чтением
ПОЛУЧИТЬ ПОДАРОКЧитайте также
Глава 2. Потоки энергии и «самоорганизация» структур
Глава 2. Потоки энергии и «самоорганизация» структур Все обменивается на огонь и огонь на все, как на золото — товары, на товары — золото. Гераклит Несмотря на внешнее разнообразие, для большинство процессов переноса (тепла, вещества, электричества) имеются общие
Глава 3. Солнце — главный источник энергии для поверхности Земли
Глава 3. Солнце — главный источник энергии для поверхности Земли О солнце, ты живот и красота природы, Источник вечности и образ божества! Тобой живет земля, жив воздух, живы воды, Душа времен и вещества! А. П. Сумароков Из большого числа возможных источников энергии,
Модель миграции В-лимфоцитов памяти: приложения для генов «домашнего хозяйства»
Модель миграции В-лимфоцитов памяти: приложения для генов «домашнего хозяйства» Могут ли соматические клетки проникать в репродуктивные органы и, таким образом, вступать в тесный физический контакт с половыми клетками? По мнению Гарри Ротенфлу роль такого «связника»
Приложения
Приложения Рабочая таблица по уходу за клематисами
Приложения
Приложения Приложение 1 Нижеследующий документ, появившийся в журнале «Nature» 16 сентября 1939 г., представляет собой совместное заявление самых видных американских и британских биологов того времени (некоторые из них Нобелевские лауреаты) и получил широкую известность как
Прилив энергии
Прилив энергии Потеря аппетита и бессонница напрямую связаны с другим удивительным признаком страсти — переполняющей человека энергией. Как сказал антропологу юноша с острова Мангайя в южной части Тихого океана, когда он думает о своей любимой, «он будто подпрыгивает
Приложения
Приложения Приложение 3 Приложение 5 Приложение 6 Приложение 7 Приложение
ПРИЛОЖЕНИЯ
ПРИЛОЖЕНИЯ Приложение 1 Соотношение возраста собаки и человека, годы (Kirk R.W., Bistner S.I.,
13. ПРИЛОЖЕНИЯ
13. ПРИЛОЖЕНИЯ ИНСТРУКЦИЯ РКФ О РЕГИСТРАЦИИ ПОТОМСТВА, ПОЛУЧЕННОГО ОТ ИСКУССТВЕННОГО ОСЕМЕНЕНИЯ СОБАК Специалист, занимающийся сбором, замораживанием, хранением. осревогскок спермы н осеменением собак замороженной спермой должен иметь договор о сотрудничестве с РКФ и
ГЛАВА 13 Приложения
ГЛАВА 13 Приложения Соотношение возраста собаки и ее хозяина Календарь повязанной суки На следующих трех страницах помещены три части этого календаря. Эти страницы нужно аккуратно вырезать и склеить друг с другом по чистым полям для склейки так, чтобы совместились
Приложения
Приложения Организация работы над учебными проектами Учебный проект – организационная форма целенаправленной учебно-исследовательской работы (деятельности), которая ориентирована на достижение конкретного результата по решению какой-либо значимой (актуальной)
Приложения
Приложения Это не первая моя книга на тему бессмертия. В предыдущих работах я тоже давал как можно проще известные положения химии и физики, учитывая, что читатели, скорее всего, начисто забыли их, давал не вошедшие в книгу факты, которые не потеряли своего значения и