Глава 24. ГДЕ СХОДЯТСЯ ВСЕ ПУТИ
Глава 24.
ГДЕ СХОДЯТСЯ ВСЕ ПУТИ
Последние четыре главы были посвящены, тем или иным образом, процессам, связанным с катаболизмом глюкозы — сначала до молочной кислоты путем анаэробного гликолиза, потом — до углекислоты и воды путем цикла Кребса. Однако нельзя сказать, чтобы это были единственные способы, которыми организм может создавать высокоэнергетические связи для хранения химической энергии.
К примеру, еще в начале 50-х годов XX века британский биохимик Ф. Диккенс с коллегами смогли показать, что во многих тканях растений и микроорганизмов, а иногда — и животных, глюкоза расщепляется на трехуглеродные соединения не путем анаэробного гликолиза, а каким-то другим путем. Этот другой путь сходится с обычным гликолизом где-то на полдороге. Он проходит через этапы пяти- и четырехуглеродных Сахаров и не является анаэробным. При нем используется и дыхательная цепочка, и атмосферный кислород.
Наличие такого запасного пути важно в двух отношениях. Во-первых, если деятельность основного механизма окажется временно нарушенной, то организм не останется совсем без ресурсов — он сможет просуществовать за счет запасного механизма.
Во-вторых, при любом катаболическом механизме производится не только энергия, но и ряд промежуточных веществ. Эти промежуточные вещества организм может впоследствии использовать как строительные материалы для процессов анаболизма. К примеру, различные промежуточные вещества, участвующие в цикле Кребса, могут служить основой для формирования некоторых аминокислот. Достаточно прибавить к щавелево-уксусной кислоте аминогруппу, и она станет аспарагиновой кислотой; прибавить аминогруппу к альфа-кетоглутаровой кислоте — она станет глютаминовой кислотой (именно поэтому наличие этих двух кислот в пище не является обязательным).
Так что разумная избыточность механизмов обмена веществ имеет свои преимущества, поскольку предоставляет организму большее количество строительных материалов. Упомянутый выше «обходной путь» приводит к созданию нескольких пятиуглеродных Сахаров и родственных им веществ. Их можно использовать для образования необходимых огромных молекул нуклеиновых кислот, чьей важной составляющей являются именно пятиуглеродные сахара.
Анаэробный гликолиз и реакции цикла Кребса тем не менее являются главными путями катаболизма глюкозы, и рассматривать альтернативные варианты более подробно сейчас не имеет смысла.
Однако приведенное в предыдущих главах описание катаболизма все же неполно. До сих пор я рассуждал только о катаболизме глюкозы. Но в главе 19 я указал, что жиры и белки, особенно последние, тоже являются важными источниками энергии. Что же происходит с ними? Явно ведь не то же самое, что с углеводами?
Давайте посмотрим.
В течение первого десятилетия XX века, когда Харден и Янг только начинали изучать промежуточный катаболизм глюкозы, немецкий биохимик Франц Кнооп взялся за решение той же задачи в отношении жиров.
Кнооп использовал технологию кормления собак строго определенной пищей с последующим изучением мочи. Однако ученый сумел серьезно улучшить эту методику. Если бы он просто кормил собак жирами и получал на выходе воду и углекислоту, то было бы совершенно непонятно, из жиров ли создана полученная молекула воды или углекислоты или из чего-то еще. Молекулы жира надо было как-то пометить, чтобы ее составные части можно было потом опознать, после того как они пройдут всю цепочку обмена веществ организма животного. Кнооп решил кормить собак фенилдеривативами различных жирных кислот.
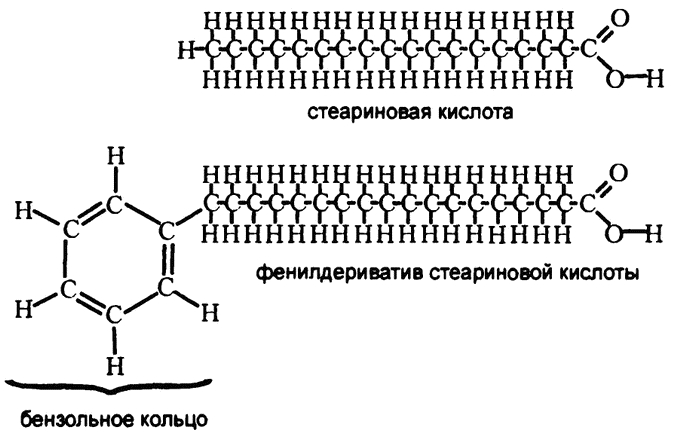
Рис. 65. Фенилдериватив жирной кислоты
Разрешите пояснить. Во-первых, жирные кислоты — это основной продукт расщепления жиров. Их молекулы состоят из длинных углеводных цепочек, обычно по шестнадцать—восемнадцать атомов углерода длиной, где один атом углерода на одном из концов цепочки входит в карбоксильную группу. В качестве примера распространенной восемнадцатиуглеродной кислоты можно назвать стеариновую кислоту. Если такое вещество подвергнуть обработке таким образом, чтобы к противоположному от карбоксильной группы концу присоединилось бензольное кольцо, то в результате получится именно фенилдериватив, изображенный на рис. 65.
Фенилдериватив жирной кислоты — это вещество, которое организм обработать не способен. Ни у собаки, ни у человека не существует ферментов, с помощью которых можно было бы разорвать само бензольное кольцо или отщепить его от углеродной цепи. Ткани организма собаки могут катаболизировать всю остальную часть жирной кислоты, но от бензольного кольца и того фрагмента жирной кислоты, к которому бензольное кольцо прикреплено, избавиться можно только выбросив его из организма с мочой. Соответственно, Кнооп мудро рассудил, что если в моче обнаружатся соединения, содержащие бензольные кольца, то это и будут остатки использованной организмом собаки жирной кислоты.
Таким образом, пометив жирные кислоты, Кнооп создал первый в мире «маркер» вещества (то есть пометку на веществе, с помощью которого можно проследить его дальнейшую судьбу). Перед тем как приступить к описанию полученных Кноопом результатов, я вкратце обрисую дальнейшую историю маркеров, поскольку эта технология оказалась крайне важной для развития биохимии в целом.
Напрашивается возражение, гласящее, что Кнооп, дескать, пичкает организм подопытного животного неестественными для того пищевыми веществами, «нефизиологичными». Пусть они нетоксичны и на здоровье организма их прием никак не отражается, но откуда у исследователя может быть уверенность, что само присутствие маркера никак не повлияло на способ обработки молекулы организмом? А вдруг бензольное кольцо подавило действие какого-нибудь важного фермента? Короче говоря, откуда мы знаем, что эти вещества следуют теми же метаболическими путями, что и нормальная, естественная пища?
Идеально было бы придумать такой маркер, чтобы организм не мог отличить помеченное вещество от непомеченного, а экспериментатор — мог. Казалось бы, нельзя требовать от мироздания слишком многого, но лет через десять после экспериментов Кноопа произошел один из самых счастливых случаев в истории науки — были открыты изотопы.
В 1913 году британский физик Фредерик Содди показал, что атомы одного элемента могут быть не абсолютно похожи. Да, все они имеют одинаковое количество протонов ядра, и такое же количество электронов на орбите, и одинаковые химические свойства (см. главу 17). Однако число нейтронов в ядрах разных атомов одного и того же вещества может различаться, а значит — и их атомный вес. Такие атомы называют «изотопами» одного и того же элемента.
Так, например, одни атомы углерода имеют ядро, содержащее шесть протонов и шесть нейтронов. Атомный вес такого атома — 12. Ядро других содержит шесть протонов и семь нейтронов и имеет атомный вес 13. На бумаге обозначение этих изотопов будет выглядеть как C12 и С13. Каждый из них будет иметь те же шесть электронов — два во внутренней оболочке и четыре во внешней; каждый будет участвовать в одних и тех же реакциях в организме. Организм не различает изотопов.
Затем в 1919 году британский физик Фрэнсис Уильям Астон изобрел масс-спектрограф — устройство, искажающее траекторию полета ионов в магнитном поле. Чем тяжелее ион, тем меньше искажается его траектория. Таким образом, стало возможно отделить C12 от С13 в случае, когда они присутствуют в одном и том же наборе. Поскольку траектория их полета искажается в разной степени, то и конечная точка попадания на фотопластину у этих двух изотопов оказывается разной, и по окончании сортировки можно подсчитать сравнительную частоту их наличия по степени потемнения пластины. Так было установлено, что в природном угле соотношение C12 к С13 составляет 989 к 11.
Теперь предположим, что пища для лабораторных животных изготавливается только с использованием С13, без единого атома C12. Для организма разницы нет. Поэтому он подвергнет вещество, содержащее С13, той же обработке, какой подверг бы и вещество, содержащее C12. Это вещество будет превращено организмом в другое, тоже с аномально высоким содержанием С13; в итоге понятно, что любое вещество с аномально высоким содержанием С13 всегда окажется происходящим от исходного питательного продукта, помеченного изотопом. Наличие аномального количества изотопа определяется экспериментатором путем сжигания вещества, получения углекислого газа и прогона этого газа через масс-спектрограф. По результатам спектрографии ученые уже могут строить предположения о метаболических механизмах, действующих в тканях живых существ.
Легко сказать, да трудно сделать. Для этого сначала надо набрать изотопов (которые встречаются в природе, как уже говорилось, очень редко), создать питательные вещества с их участием, потом выделить из организма другие вещества в чистом виде и в конце концов провести очень непростую процедуру масс-спектрографий. Из-за всех этих трудностей масс-спектрография так и не получила широкого распространения до 1932 года, когда американский химик Гарольд Юри открыл дейтерий, или тяжелый водород.
Ядро обычного водорода состоит только из протона, так что его атомный вес — 1, а ядро дейтерия — из протона и нейтрона, поэтому его атомный вес — 2. Эти изотопы обозначаются, соответственно, как Н1 и Н2. И если между C12 и С13 разница масс составляет 8 процентов, то между Н1 и Н2 — 100 процентов.
Из такой колоссальной разницы масс между водородом и дейтерием можно извлечь определенные преимущества. Более тяжелые изотопы участвуют в тех же реакциях, что и более легкие, но с меньшей скоростью. Для всех остальных элементов, кроме водорода, разницей в скорости вполне можно пренебречь; однако в случае с водородом уже заметно, что реакции с участием дейтерия протекают медленнее. Перегонка и электролиз воды, в состав которой входит не водород, а дейтерий, происходят заметно медленнее. Если подвергать перегонке или электролизу большое количество воды, то ближе к концу процедуры оставшаяся часть воды будет представлять собой высокую концентрацию воды тяжелой. Из такой тяжелой воды можно изготавливать дейтерийсодержащие продукты. Ну и отделять дейтерий от водорода в спектрографе тоже сравнительно несложно.
В 1935 году американский физик немецкого происхождения Рудольф Шёнхеймер пометил жирные кислоты дейтерием, так же как Кнооп за поколение до того — бензольным кольцом. Позже, по мере развития технологий, Шёнхеймер использовал для пометки аминокислот тяжелый азот, N15 (обычный азот — N14). В результате своих экспериментов он сумел доказать, что организм ведет чрезвычайно активную химическую деятельность. Даже в тех ситуациях, когда никаких видимых следов активности незаметно, постоянно идут перемещения, переносы, соединения и распады, постоянно перемешиваются атомы.
Но технология маркирования изотопами по-прежнему оставалась неуклюжей и неудобной. Попав в организм, вещество, содержащее изотоп, растворялось по всему организму, и найти его потом оказывалось очень непросто.
Но выход был найден. Атомы некоторых изотопов содержат неустойчивое соединение протона с нейтроном. Из таких атомов с известными скоростями и в известном количестве испускаются различные субатомные частицы. К примеру, С14, ядро которого состоит из шести протонов и восьми нейтронов, испускает энергетические частицы такими темпами, что половина С14 превращается в N14 (самый распространенный вариант азота) за пять тысяч лет.
Такое явление называется радиоактивностью. Если в пищевые вещества включить радиоактивный атом С14, то с точки зрения обмена веществ он будет проявлять все те же свойства, что и обычные стабильные изотопы C12 или С13, но по маркеру из энергетических частиц экспериментатор всегда сможет его найти. С14 можно отловить даже в том случае, если его концентрация крайне мала.
Оставалось только придумать способ производства самих радиоактивных изотопов в достаточном количестве. До Второй мировой войны приходилось полагаться только на естественно встречающиеся в природе радиоактивные изотопы — а это были по большей части изотопы атомов, не участвующих в пищевой цепочке животных. Но после окончания войны практически ни одно исследование метаболизма уже не велось без использования веществ, помеченных радиоактивными изотопами С14, Н3, Na24 и так далее. Теперь работу биохимика невозможно представить без них.
Но вернемся же к Кноопу и его первым в мире опытам с молекулярными маркерами…
Предоставляя собакам с пищей маркированные жирные кислоты с четным количеством атомов углерода, ученый выделял из мочи животных бензольное кольцо в сочетании с группой, включающей в себя два атома углерода, — такое вещество называется фенилуксусной кислотой. Кислота эта, в свою очередь, крепилась к молекуле глицина. Если же на входе подавались кислоты с нечетным числом атомов углерода, то на выходе получалось бензольное кольцо, к которому крепился только один атом углерода («бензойная кислота»), и она тоже крепилась к молекуле глицина (рис. 66). Организм часто добавляет глицин к молекуле, которую надо вывести из организма, чтобы она легче выводилась через почки, так что Кнооп не стал обращать на глицин внимания, а сосредоточился на фенилуксусной и бензойной кислотах.
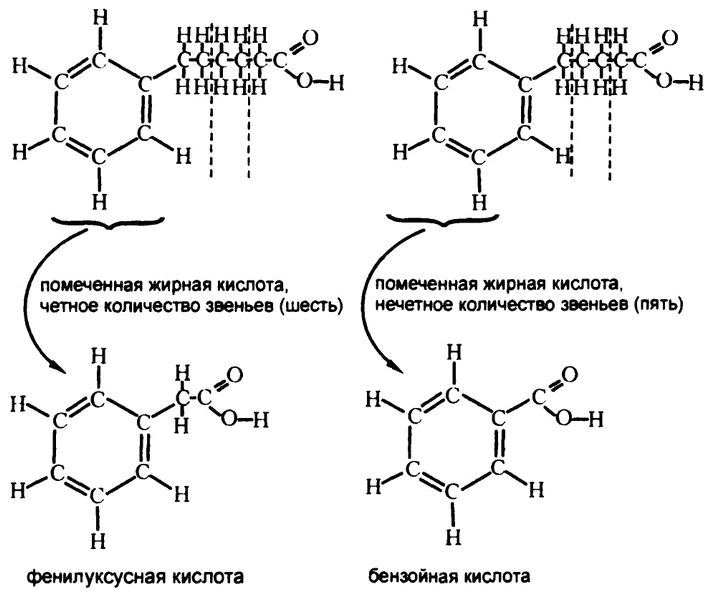
Рис. 66. Катаболизм жирных кислот
Кнооп объяснил полученные результаты тем, что длинная цепочка жирной кислоты разрезается на блоки по два атома углерода, начиная с карбоксильной группы. Оторвать углерод от бензольного кольца организм не может, поэтому если общее количество звеньев цепочки оказывается четным, то прикрепленными к бензольному кольцу остаются два звена, если нечетным — то одно.
Такое предположение казалось тем более логичным, что известно, что все жирные кислоты всех жиров живых тканей состоят из четного числа атомов углерода — значит, двухуглеродные блоки по какой-то причине естественны для организма, и именно их он использует при строительстве собственных жирных кислот. В природе очень часто встречаются восемнадцати- и шестнадцатиуглеродные жирные кислота, а семнадцатиуглеродных не встречал пока еще никто (те жирные кислоты с нечетным числом атомов углерода, которые Кнооп скармливал собакам, не в счет, они были искусственно синтезированы в лаборатории).
Два года спустя немецкий биохимик Густав Эмбден внес в систему еще одно усовершенствование. Он обнаружил, что при обработке жирной кислотой тканей печени в растворе появляется цепочка из четырех атомов углерода — ацетоуксусная кислота:
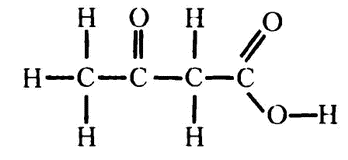
Это вещество вместе с рядом других входит в группу «кетонных тел», названных так из-за присутствия кетогруппы С=О. Ацетоуксусная кислота — пример жирной кислоты, от которой начал отщепляться двухуглеродный отрезок, как я сейчас объясню, и это утвердило Кноопа в его предположении о «нарезании» организмом двухуглеродных отрезков.
Кетонные тела не встречаются в каких-либо значительных количествах ни в крови, ни в моче здоровых животных, несмотря на то что печень производит их, будучи изолированной. А вот в крови и моче диабетиков — встречается! Поскольку диабет — это расстройство в первую очередь углеводного метаболизма, то его связь с производством кетонных тел — продуктов метаболизма жиров — демонстрирует связь между этими двумя метаболическими механизмами.
Несмотря на столь давний интерес ученых к проблеме, подробности метаболизма жиров оставались неизвестными до тех пор, пока не была разработана вышеописанная технология изотопного маркирования. Лишь в 1951 году биохимики получили возможность писать формулы жирового метаболизма хоть с какой-то степенью уверенности.
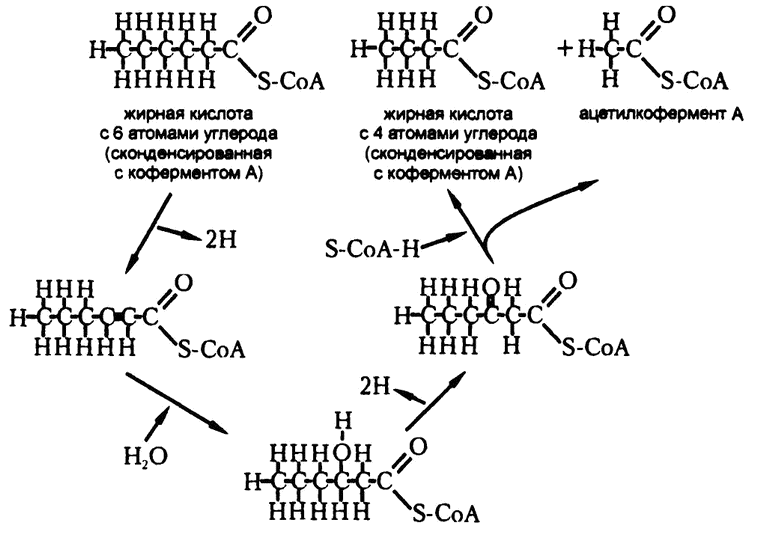
Рис. 67. Цикл окисления жирной кислоты
В свете сегодняшнего знания мы можем сказать, что Кнооп был прав: действительно, углеродная цепочка жирных кислот «нарезается» организмом на фрагменты по два атома углерода, начиная с карбоксильного конца. Только надо внести два уточнения. Во-первых, для того, чтобы подвергнуться катаболизму, жирная кислота должна конденсироваться с коферментом А. Во-вторых, сам процесс отрезания двух атомов углерода происходит в четыре этапа (рис. 67).
Первый этап — дегидрогенизация. От каждого из двух атомов углерода, соседствующих с карбоксильной группой, отщепляется по атому водорода. Второй этап — гидратация. К веществу добавляется вода таким образом, что гидроксильная группа присоединяется ко второму атому углерода карбоксильной группы (этот атом называется «бета-углерод», соответственно, вся реакция имеет название «бета-оксидация»). На третьем этапе удаляются еще два атома водорода, так что бета-углерод становится частью кетогруппы. Если посмотреть сейчас на формулу ацетоуксусной кислоты, то станет ясно, что она — продукт именно этого, третьего, этапа. На четвертом этапе два атома углерода отщепляются одновременно в составе ацетилкофермента А, и от цепочки остается новая, укороченная на два атома углерода жирная кислота, которая конденсируется с коферментом А и отправляется на следующую итерацию цикла.
Полное название этого цикла — «цикл окисления жирной кислоты». На самом деле это скорее спиралеобразный, чем циклический, процесс, поскольку нельзя сказать, что в конце мы приходим к тому же, что имели в начале, поскольку с каждой итерацией жирная кислота становится короче на два атома углерода. Таким образом, восемнадцатиуглеродная жирная кислота, например стеариновая, в конце концов превращается в девять молекул ацетилкофермента А.
После формирования ацетилкофермента А он вступает в соединение с щавелево-уксусной кислотой для образования лимонной кислоты и входит, таким образом, в цикл Кребса, описанный в предыдущей главе. Не важно, сформирован ли ацетилкофермент А из жирной кислоты или путем катаболизма глюкозы; организм не может определить происхождение вещества (рис. 68).
Ацетилкофермент А тоже можно рекомбинировать с получением в итоге жирной кислоты. Для этого надо обратить цикл окисления жирных кислот. То есть две молекулы ацетилкофермента А объединяются для получения ацетоацетилкофермента А, затем добавляются два атома водорода, удаляется вода, добавляются еще два атома водорода — и вот мы получили четырехуглеродную жирную кислоту, конденсированную с коферментом А. Эту цепочку можно продолжать сколько угодно, добавляя каждый раз по ацетилкоферменту А.
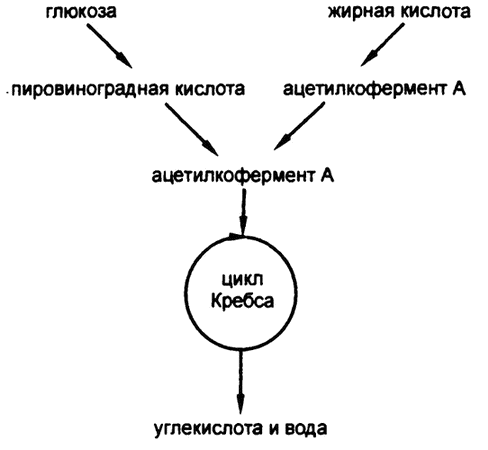
Рис. 68. Соединение путей катаболизма углевода и жира
Получается, что ацетилкофермент А и есть тот самый общий строительный «кирпичик», наличие которого я логически обосновал в главе 19, рассуждая о том, как крахмалистая пища переводится в жир, а нормальный уровень глюкозы в крови поддерживается за счет расщепления жира.
Конечно, и в метаболизме жирных кислот, как и в метаболизме глюкозы, важное место занимает дыхательная цепь. При каждой итерации цикла окисления жирных кислот происходит две дегидрогенизации. Первая катализируется флавоферментом, а вторая — пиридин-ферментом. Можно считать, что в первом случае производится две молекулы АТФ, а во втором — три, так что при каждой итерации цикла производится в целом пять молекул АТФ путем окислительного фосфорилирования.
В случае, скажем, стеариновой кислоты, в состав которой входят восемнадцать атомов углерода, происходит восемь итераций цикла (понятно, почему не девять? Попробуйте разрезать полоску бумаги на 9 частей, и увидите, что потребуется совершить всего 8 разрезов), и при этом производится девять молекул ацетилкофермента А.
То есть при переводе молекулы стеариновой кислоты в девять молекул ацетилкофермента А получается 8 умножить на 5, то есть 40 молекул АТФ. Если посмотреть на схему цикла Кребса, приведенную на рис. 64, то видно, что в этом цикле из ацетилкофермента А производится 12 молекул АТФ. Соответственно, если 9 молекул ацетилкофермента А умножить на 12, получим 108 молекул АТФ.
Таким образом, в процессе перехода от стеариновой кислоты к углекислоте и воде образуется не менее 40+108, то есть 148 молекул АТФ. Но это еще не все. Типичная молекула жира, например стеарин, состоит из трехуглеродного спирта, глицерина, в соединении с тремя молекулами стеариновой кислоты. Каждая из трех молекул стеариновой кислоты послужит формированию 148 молекул АТФ, то есть всего образуется 3 x148 = 444 молекулы АТФ. Глицерин же будет превращен в пировиноградную кислоту, что само по себе даст еще 15 молекул АТФ — итого организм получит 459 молекул АТФ в результате катаболизма каждой молекулы стеарина.
Идем дальше. При переходе одного грамма жира в углекислоту и воду высвобождается 9 килокалорий тепла (см. главу 14). Один моль стеарина весит 891 грамм, соответственно, при переходе одного моля стеарина в углекислоту и воду высвобождается 891 х 9 = 8019 килокалорий. Путем образования 459 молекул АТФ запасается 459 х 8 = 3672 килокалории химической энергии. Значит, эффективность процесса — 3672: 8019 = чуть более 45 процентов. Это превосходный результат.
Но если посмотреть на рис. 64, видно, что ацетилкофермент А попадает в цикл Кребса путем соединения с щавелево-уксусной кислотой. А щавелево-уксусная кислота производится в достаточном количестве только путем углеводного метаболизма (до какой-то степени — белкового метаболизма), но никак не метаболизма жиров.
Если в пище много жиров, но мало углеводов, ацетилкофермента А производится много, но щавелево-уксусной кислоты — мало. Ацетилкофермент А начинает накапливаться, конденсироваться сам с собой, образовывая ацетоуксусную кислоту, и в организме образуется повышенная концентрация кетонных тел, находящая выражение в состоянии, известном как «кетоз».
Такая диета, богатая жирами, но бедная углеводами, называется «кетогенной», потому что способствует повышению концентрации кетонных тел. Голодание — тоже кетогенный процесс, потому что все углеводородные запасы организм исчерпывает за день, а дальше начинаются процессы извлечения энергии из жиров. Если печень обрабатывать жирной кислотой, не добавляя при этом углеводов, то, по наблюдениям Эмбдена, тоже начинается кетоз.
Ни голодание, ни кетогенные диеты обычно не приносят большого вреда, поскольку человеку свойственно избегать и первого, и второго. Но даже если обстоятельства вынуждают человека к таким условиям, то даже небольшое количество переваренных углеводов произведет корректирующий антикетогенный эффект.
Вот при диабете кетоз становится действительно опасным. Если болезнь не лечить, то развивается хроническое расстройство углеводного метаболизма, приводящее ко все более жесткой нехватке щавелево-уксусной кислоты и, соответственно, прогрессирующему кетозу. Именно токсическое действие кетоновых тел обычно и приводит к коме и смерти диабетика.
Строго говоря, не все жиры имеют своим основным предназначением производство энергии. Те жиры, что я обсуждал в этой главе, относятся к «простым» жирам, состоящим из глицерина и жирных кислот. Существуют также «сложные» жиры, в состав которых входят еще и фосфатная группа и одна из нескольких азотсодержащих групп. А еще есть такие вещества, как холестерин, обладающие свойствами жиров, но имеющие совершенно другое молекулярное строение.
У этих последних в организме есть особые функции. Из фосфатсодержащих жиров состоит мембрана митохондрий, служащая, как я уже писал, полем деятельности для различных ферментов, катализирующих реакции цикла Кребса и дыхательной цепочки. Что же касается холестерина, то из него состоит большая часть жировой оболочки нервов.
Сложные жиры и холестерин содержат не меньше энергии, чем простые жиры, но их роль в клеточном механизме настолько велика, что их энергозапасы не расходуются даже при крайних стадиях истощения. Я не привожу здесь подробного рассмотрения этих крайне важных веществ только потому, что они не задействованы в процессах энергообмена.
Теперь давайте вернемся к белкам, попадающим после процесса переваривания в организм в виде аминокислот. Перед тем как использовать их для производства полезной энергии, надо сначала убрать из них азот.
В 1773 году французский химик Руэль (учитель Лавуазье) обнаружил в моче азотистое соединение и назвал его, естественно, мочевиной. Когда в начале XIX века химики стали изучать строение белков, мочевина была сразу признана «тем самым» веществом, посредством которого организм избавляется от азотистой составляющей белков.
Формула мочевины —
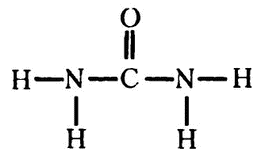
Ее короткая запись — NH2CONH2. Получилось так, что именно с мочевиной оказались связаны два самых замечательных открытия в биохимии. Она стала первым органическим веществом, которое удалось синтезировать из неорганического материала (см. главу 13), и катализирующий ее распад фермент был кристаллизован первым (см. главу 15).
В XIX веке способ образования мочевины в организме оставался неизвестен. Однако уже в первое десятилетие XX века, когда Харден и Янг, с одной стороны, и Кнооп — с другой, начали продвижение в тайны промежуточных этапов метаболизма, немецкий биохимик Альбрехт Коссель вместе с английским коллегой Генри Дэкином пошли в такое же наступление на секреты промежуточных этапов белкового метаболизма.
Они обнаружили фермент, названный ими «аргиназа», действие которого заключалось в катализировании гидролиза аминокислоты аргинина. При этом с помощью реакции, изображенной на рис. 69, образовывалась и мочевина. Кусок молекулы аргинина, остающийся по завершении процесса, — это аминокислота орнитин, которая встречается только как промежуточный продукт образования мочевины, и частью белковых молекул не является.
Значение аргиназы подчеркивают несколько фактов. Мочевина производится в печени (если удалить у животного печень, производство мочевины прекращается), и наивысшая концентрация аргиназы наблюдается тоже в печени. Аргиназа встречается в тканях всех животных, в чьем организме производится мочевина, например амфибий или млекопитающих. А вот в тканях животных, в чьем организме производятся другие азотсодержащие продукты — птиц, например, или рептилий, — она не встречается. Понятно, что аргинин и аргиназа участвуют в каком-то механизме, с помощью которого живые ткани синтезируют мочевину.
Организм человека производит в день от десяти до тридцати пяти граммов мочевины, не истощая при этом запасов аргинина. Значит, они возобновляются путем синтеза аргинина из орнитина.
Это стало окончательно ясно в 1932 году благодаря работам Кребса (того самого, впоследствии — первооткрывателя цикла Кребса) и его коллеги К. Хенселейта с образцами печеночной ткани. Они обнаружили, как и ожидалось, что добавление аргинина стимулирует производство мочевины. Такой же эффект оказывало и добавление орнитина, и этот факт утвердил ученых в мысли, что печень производит из орнитина аргинин. В конце концов они обнаружили, что еще одно вещество, сходное по строению с аргинином — цитруллин, — тоже стимулирует производство мочевины.
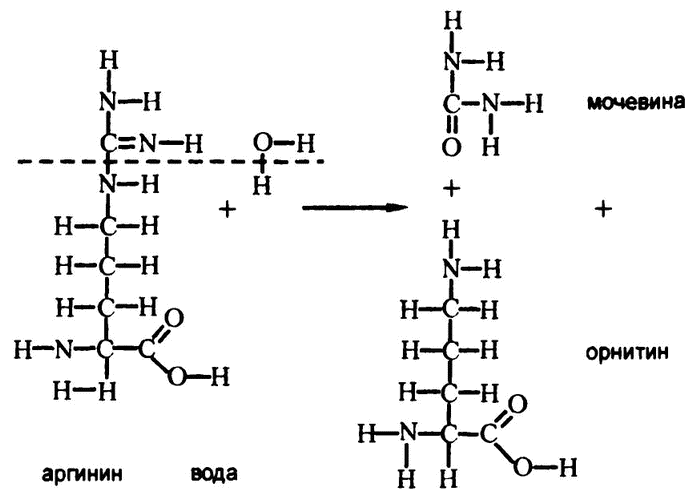
Рис. 69. Гидролиз аргинина
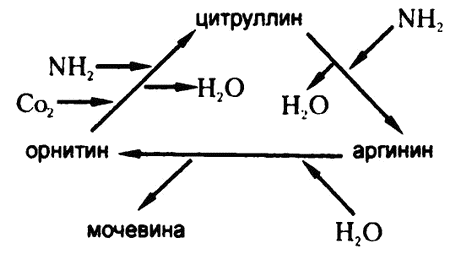
Рис. 70. Цикл образования мочевины
В качестве объяснения Кребс и Хенселейт составили схему, которую принято сейчас называть «циклом образования мочевины» — она приведена на рис. 70. Эта схема показывает, что орнитин, цитруллин и аргинин постоянно синтезируются заново. Смотрите, при превращении орнитина в цитруллин организм добавляет одну молекулу аммиака и одну молекулу углекислоты, а при превращении цитруллина в аргинин — еще одну модекулу аммиака. Что же касается воды, то одна ее молекула добавляется организмом на этапе гидролиза аргинина, но на каждом из двух этапов одна молекула воды из системы извлекается, так что в целом можно говорить об извлечении одной молекулы воды. И в конце концов при гидролизе аргинина извлекается одна молекула мочевины. Вот как выглядит запись всей реакции в целом:
2NH3 + СO2 ? NH2CONH2 + H2O.
Конечно, приведенная схема цикла образования мочевины сильно упрощена. Никто из выдвинувших ее на самом деле, конечно, не считал, что на входе подается молекула аммиака как таковая. Аммиак — очень ядовитое вещество, и в организме в чистом виде встречаться не может. Однако только в 50-х годах XX века, когда начали проводить эксперименты с изотопным маркированием, стали известны подробности этих реакций.
Выяснилось, что азотсодержащие группы добавляются в виде аминогрупп, получаемых из аминокислот. При переходе от орнитина к цитруллину глютаминовая кислота (одна из наиболее распространенных аминокислот) отдает и аминогруппу, и эквивалент углекислоты. В этом случае она должна использовать высокоэнергетическую фосфатную связь, поручаемую из АТФ. При переходе от цитруллина к аргинину аминогруппу отдает другая аминокислота, аспарагиновая, и снова за счет АТФ.
То есть производство мочевины — это энергоемкая реакция. На каждой итерации цикла потребляется по две молекулы АТФ.
Не стоит считать, что единственными аминокислотами, способными отдавать аминогруппу на производство мочевины, являются глютаминовая и аспарагиновая кислоты. В 30-х годах XX века, работая с изотопом N15, о чем я рассказывал в этой же главе, Шёнхеймер установил, что если дать на входе один вид аминокислоты, помеченный изотопом, то вскоре изотопы окажутся в составе аминокислот всех видов, за исключением лизина. Естественно, напрашивается вывод, что аминогруппа легко передается от одного вещества к другому. Были обнаружены и ферменты, катализирующие подобные реакции (трансаминазы), и установлены подробности переносов, получивших название «трансаминирование» (от «транс» — перенос и «амино»). После трансаминирования аминогруппу NH2 заменяет связанный двойной связью кислород (рис. 71).
Трансаминазы представляют собой пример ферментов, использующих в качестве кофермента пиридоксальфосфат, в котором содержится группа атомов, организмом не синтезируемая в принципе. Так был обнаружен еще один витамин группы В, пиридоксин.
Итак, движение аминогрупп осуществляется от аминокислот в целом к глютаминовой кислоте и аспарагиновой кислоте, а от них уже в цикл производства мочевины. Те аминокислоты, в состав которых входит лишний атом азота или атом серы, избавляются от них с помощью механизмов, которые я здесь не буду описывать. Достаточно будет сказать, что, так или иначе, все атомы азота и серы устраняются и остается только «углеродный скелет».
Этот углеродный скелет может представлять собой одно из тех веществ, с которыми мы уже сталкивались при описании катаболизма углеводов или белков. К примеру, когда аланин подвергается трансаминации, он теряет аминогруппу и приобретает связанный двойной связью кислород, вследствие чего образуется пировиноградная кислота.
С помощью кофермента А пировиноградная кислота избавляется от гидроксильной группы обычным способом, и образуется ацетилкофермент А, который уже может вступить в цикл Кребса. Для организма нет разницы, образована ли пировиноградная кислота из глюкозы или из аланина. И в том и в другом случае это одна и та же пировиноградная кислота, с одним и тем же энергетическим содержанием. Другие аминокислоты, например валин, серии, треонин и цистеин, тоже преобразуются, несколько более длинным путем, в пировиноградную кислоту.
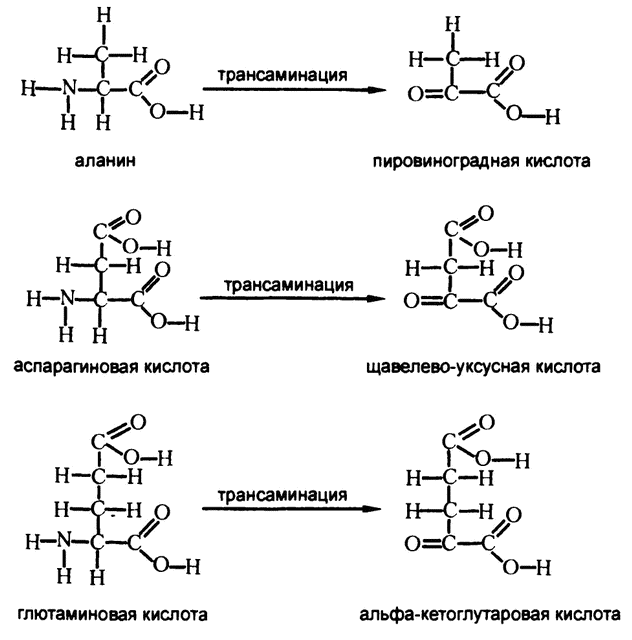
Рис. 71. Трансаминация
Несколько аминокислот, например лизин, подвергаются изменениям, в ходе которых превращаются в ацетоуксусную кислоту и далее — в ацетил-кофермент А, после чего входят в цикл Кребса по пути катаболизма жиров.
А некоторые аминокислоты входят в цикл Кребса и сами по себе. К примеру, аспарагиновая кислота сразу после деаминации становится щавелево-уксусной кислотой, а глютаминовая кислота — альфа-кетоглутаровой кислотой. Оба этих вещества являются промежуточными продуктами цикла Кребса и могут сразу быть приняты в него на соответствующих этапах.
На рис. 72 подведены итоги катаболических взаимосвязей белков, жиров и углеводов.
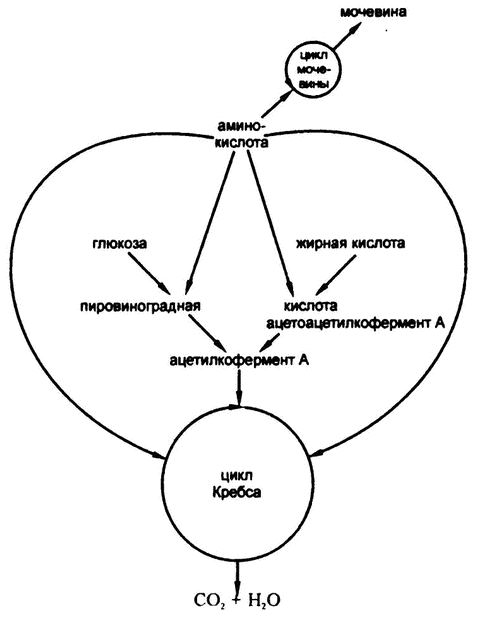
Рис. 72. Присоединение белка
Конечно, обратив различные реакции катаболизма, можно получить аминокислоты из соответствующих углеродных скелетов. К примеру, если организм пресыщен глютаминовои кислотой и в то же время наблюдается нехватка аланина, в некоторых случаях запускается процесс передачи аминогруппы от глютаминовои кислоты к пировиноградной кислоте и формирования из последней аланина. В случае обратной ситуации организм точно так же может инициировать процесс передачи аминогруппы от аланина к альфа-кетоглутаровой кислоте, а из последней — образовывать глютаминовую. Присутствие в пище аминокислот, которые могут быть синтезированы подобным образом, не обязательно.
С другой стороны, некоторые аминокислоты проходят во время реакций получения углеродных скелетов хотя бы один необратимый этап. Такие аминокислоты уже нельзя вернуть в исходное состояние с помощью каких бы то ни было промежуточных веществ. Их присутствие в пище необходимо, я приводил список этих аминокислот в главе 19.
Более 800 000 книг и аудиокниг! 📚
Получи 2 месяца Литрес Подписки в подарок и наслаждайся неограниченным чтением
ПОЛУЧИТЬ ПОДАРОКЧитайте также
Глава 4. Два пути
Глава 4. Два пути Факт или артефакт? Профессор С. Северин, узнав, что вслед за Шарфшвертом я освоил заокеанскую методику, попросил применить ее к другому объекту: вместо печени крысы надо было взять грудную мышцу голубя. План моего руководителя состоял в том, чтобы
Начало пути
Начало пути Однажды, просматривая в библиотеке биофака новые журналы, я наткнулся на короткую статью в «Нэйчер» под названием «Сопряжение окисления и фосфорилирования механизмом хемиосмотического типа». Автор П. Митчел — новое имя в биоэнергетике. И термин
ПУТИ СНАБЖЕНИЯ
ПУТИ СНАБЖЕНИЯ У берегов восточного Крыма, там, где горбится величественный горный массив Карадаг, прямо из голубых вод Черного моря поднимается грандиозная скала Золотые ворота, похожая на огромную арку, увенчанную шпилем. Старожилы окрестных городов и поселков,
2. Два пути завоевания
2. Два пути завоевания Люди создают культуры, используя податливые языки. Мы изобретаем понятные нам символы и за счет них выстраиваем коммуникационные сети, на много порядков более обширные, чем у животных. Мы завоевали биосферу и опустошили ее так, как никакой другой вид
ПУТИ СНАБЖЕНИЯ
ПУТИ СНАБЖЕНИЯ У берегов восточного Крыма, там, где горбится величественный горный массив Карадаг, прямо из голубых вод Черного моря поднимается грандиозная скала Золотые ворота, похожая на огромную арку, увенчанную шпилем. Старожилы окрестных городов и поселков,
Глава 2 От синтетической теории эволюции к эволюционной геномике: различные механизмы и пути эволюции
Глава 2 От синтетической теории эволюции к эволюционной геномике: различные механизмы и пути эволюции Пер. А. НестеровойВ этой главе мы продолжим обсуждение эволюционной биологии в период до появления геномики. Многие из обсуждаемых направлений развития не являлись
Глава 4 Препятствия на пути кислорода
Глава 4 Препятствия на пути кислорода В нормальной атмосфере гемоглобин связывает только кислород. Это значит, что на связывание кислорода не оказывают воздействия другие компоненты воздуха: азот, двуокись углерода, пары воды или аргон. Гемоглобин собирает
Глава 17. Пути метаболизма глюкозы
Глава 17. Пути метаболизма глюкозы Глюкоза является основным метаболитом и транспортной формой углеводов в организме человека и животных. Источниками глюкозы являются углеводы пищи, гликоген тканей и процесс глюконеогенеза в печени и корковом веществе почек. Для
1.4. Дыхательные пути
1.4. Дыхательные пути Гайморовы пазухи, глотка, гортань, внеторакальные (расположенные выше грудной клетки) участки трахеи и так далее, передающие поток воздуха из окружающей среды вниз к альвеолам через дыхательные ворота организма, а также нос и рот, определяются как
Начало пути…
Начало пути… Не секрет, что в нашем обществе традиционно утвердилось мнение о том, что употребление мяса естественно для человека. В связи с этим «рядовому» представителю этого общества практически не оставляется шансов узнать о последствиях связанных с его
На пути к доместикации
На пути к доместикации «Доместикация» значит «одомашнивание». Есть виды животных, прирученные человеком, которые близки к тому, чтобы стать домашними. И один из наиболее вероятных кандидатов — африканская антилопа канна. Собственно говоря, в Древнем Египте она уже была
15. В конце пути
15. В конце пути Воистину, чем больше я смотрю на творения природы, тем более готов увидеть в ней самое невероятное. Плиний Во введении к этой книге я пригласил вас, читатель, отправиться со мною в необычное путешествие. Надеюсь, теперь вы увидели своими глазами, насколько
Глава IV. Пути изучения фенофонда
Глава IV. Пути изучения фенофонда Понятие «фенофонд» родилось в нашей стране. В московской школе генетиков, возглавляемой Н. К. Кольцовым, и в ленинградской школе, возглавляемой Ю. А. Филипченко, в 20-х годах интенсивно шло обсуждение популяционно-генетических вопросов. В
Глава 3. Неисповедимы пути полового отбора
Глава 3. Неисповедимы пути полового отбора Тайны полового отбора Эволюция в направлении производства половых клеток разного размера сама создает новое селективное давление, которое способствует дальнейшей дифференциации двух морфотипов гамет. Происходит это