Глава 18. КАК РАБОТАЮТ ФЕРМЕНТЫ
Глава 18.
КАК РАБОТАЮТ ФЕРМЕНТЫ
Тот факт, что белковые молекулы имеют спиралевидную форму, удерживаемую водородными связями, объясняет не только хрупкость молекулы фермента и легкость, с которой она теряет свои свойства, но и огромную вариативность этих самых свойств.
Давайте сравним платиновый порошок и молекулу фермента. Как я уже объяснял в главе 12, каталитические способности платины, возможно, объясняются тем, что она имеет свободные валентные связи, с помощью которых притягивает к себе молекулы тех веществ, реакцию которых катализирует. Такое связывание происходит несколько активнее, если поверхность платинового катализатора имеет неправильную форму, — поскольку молекулы реагирующих веществ также имеют неправильную форму, то им может оказаться естественнее плотно прилечь там, где форма поверхности платины соответствует их собственной.
На поверхности платины возможны любые неправильности, поэтому для любой молекулы там окажется соответствующая форма. В результате множество различных молекул могут оказаться временно привязанными к поверхности платины, где они, соответственно, с готовностью вступают в реакцию с другими молекулами. Поэтому платина, как и все прочие катализаторы такого рода, способна охватывать своим действием сравнительно большое число веществ. Но у катализаторов такого рода есть и недостаток — поскольку в каждом отдельно взятом веществе они действуют далеко не на всю площадь (а только на ту ее часть, неправильность формы которой соответствует неправильности формы реагирующих молекул), то и коэффициент полезного действия таких молекул невелик. Для каждого отдельно взятого вещества большая часть поверхности катализатора оказывается неиспользуемой.
С ферментами все обстоит по-другому. Их поверхность имеет не случайную форму, а, напротив, строго упорядоченную с помощью биохимических процессов, происходящих в организме. Чередующиеся радикалы разных аминокислот, составляющих пептидную цепочку, представляют неправильности всех типов и размеров. Некоторые из радикалов, например аспарагиновая кислота или глютаминовая кислота, обладают отрицательными зарядами и будут притягивать любой положительный заряд, как только в пределах досягаемости окажется несущая такой заряд молекула. Другие же, например лизин, аргинин или гистидин, обладают положительным зарядом и будут притягивать молекулы с отрицательными. Третьи, например серии, треонин или тирозин, электрически нейтральны, зато имеют в своем составе группы, способные к образованию водородных связей.
Что же касается радикалов, не образующих связей, например валина, аланина, лейцина, изолейцина, фенилаланина и еще нескольких, то их роль в данном случае защитная; они служат барьерами для того, чтобы только молекулы строго определенной формы могли проникнуть в зону досягаемости тех радикалов, которые смогут их ухватить.
Все радикалы находятся друг к другу в строго определенном отношении, зафиксированном водородными связями, удерживающими молекулу в жесткой форме. В результате к определенному ферменту может присоединиться молекула только строго определенного вида, и только такая молекула в дальнейшем вступит в катализируемую ферментом реакцию. Молекула, подвергающаяся реакции, катализируемой ферментом, именуется субстратом этого фермента. Разумеется, любое воздействие на фермент, приводящее к разрыву водородных связей и установленных для него отношений между радикалами, приведет к потере ферментом его уникальных каталитических свойств.
У такой строгой специфичности молекулы фермента есть одно интересное следствие. Стоит лишь задуматься о химических механизмах жизни как таковой, как становится ясно, что функционирование организма зависит от бесперебойной работы тысяч различных химических реакций с участием множества веществ. Если ферменты настолько специфичны, то означает ли это, что каждый организм — да что там организм, что каждая клетка в организме! — должен быть полон различных ферментов, каждый из которых необходим для одной-единственной реакции?
Да. Так оно и есть. Хоть клетка и имеет крайне малый объем, около 0,03 мм в диаметре, она все же гораздо больше, чем молекула белка. В клетке достаточно места для тысяч различных ферментов. Более того, места хватит для тысяч молекул каждого из них.
На первый взгляд это совершенно неразумная избыточность. Но на самом деле — ровно наоборот, экономичность и эффективность такой системы выше всяких похвал.
Каждый фермент, имеющий строго определенную задачу, обладает гораздо большей полезной площадью поверхности, чем случайным образом оформленная платиновая частица. Он гораздо эффективнее.
Скорость катализируемой реакции измеряется «оборотным числом». Оборотное число — это количество молекул субстрата, ежеминутно подвергающихся реакции при заданной температуре под действием одной молекулы фермента. Фермент каталаза, катализирующий распад перекиси водорода на воду и кислород, имеет оборотное число 2 500 000 при 0 °С. Оборотные числа большинства ферментов ниже, чем каталазы, но все равно многократно выше, чем у катализаторов неферментного происхождения.
Предположим, что различные ферменты в некотором порядке распределены по клетке (а так оно и есть, как мы увидим позже), и тогда движущиеся по клетке субстраты каждую секунду систематически улавливаются и подвергаются преобразованиям.
Кроме того, наличие множества специализированных катализаторов, а не нескольких общих, предоставляет организму возможность тончайшего управления клеточной химией. Уменьшая или увеличивая число молекул того или иного фермента в клетке, можно ускорять или замедлять ход той или иной реакции — одной из тысяч, и только ее.
Если в живом организме ежесекундно проходят тысячи различных реакций взаимодействия, независимо контролируемые тысячами различных ферментов, то выходит, что даже самые примитивные формы жизни устроены сложнее, чем самые сложные рукотворные механизмы.
Факт в том, что живая клетка состоит из гораздо большего количества движущихся деталей, так сказать, чем любое творение рук человеческих, и лишь микроскопически малый размер этих деталей позволяет нам свысока утверждать, что клетка — это «всего лишь» микроскопическая капелька. А если мы говорим о целых организмах, состоящих из триллионов клеток, то тут сложность всей конструкции возрастает до неописуемых величин. Нелепы были бы попытки сравнить ящерицу с камнем (разве что по параметру физического размера). Поэтому я даже не буду задерживаться на сравнивании человека со звездой — понятно, что сравнение будет не в пользу звезды.
Разумеется, в основе вышеприведенного анализа каталитической деятельности ферментов лежит представление о том, что субстрат вступает с ферментом в физический контакт и образует некий промежуточный комплекс «субстрат-фермент». Сложно избежать этого допущения, потому что иначе пришлось бы предположить, что фермент оказывает на молекулу некое бесконтактное воздействие — а ученые не любят бездоказательной мистики.
К тому же имеются убедительные, хоть и косвенные, свидетельства в пользу такого предположения. К примеру, предположим, что в раствор фермента добавили немного субстрата. Реакция начинается сразу же, и химики могут определить скорость, с которой исчезает субстрат или появляется итоговое вещество реакции, — так они получают скорость реакции. Если в тот же раствор фермента добавить больше субстрата — скорость реакции будет выше. Можно воспользоваться такой аналогией: если в супермаркет запустить больше покупателей, то они начнут выносить больше товаров. Если одномоментно удвоить количество покупателей в супермаркете, то логично ожидать, что и товары с полок будут исчезать в два раза быстрее. Вот по тому же принципу удваивается и скорость реакции, если удвоить концентрацию субстрата.
Однако все это верно лишь до определенного момента. По достижении некоторой концентрации субстрата скорость реакции достигает максимума и выше уже не поднимается, сколько субстрата ни добавляй (рис. 32). Аналогия с супермаркетом здесь не теряется — потому что скорость раскупания товара тоже не может возрастать вечно. Как только забиваются кассы, дальнейшее увеличение количества покупателей приводит только к удлинению очередей.
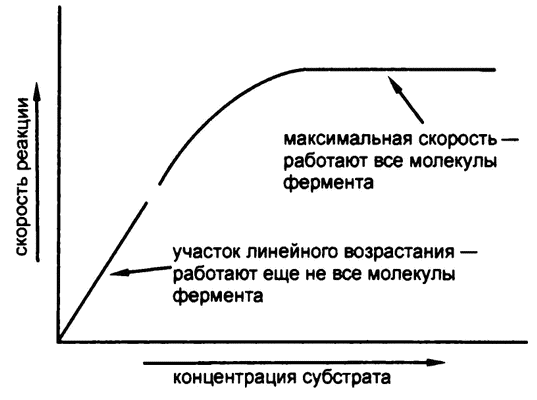
Рис. 32. Зависимость скорости реакции от концентрации субстрата
Точно так же, когда концентрация субстрата достигает такого значения, что каждая молекула фермента работает на полную мощность, дальнейшее добавление субстрата приводит только к увеличению, так сказать, очередей его молекул, стоящих в очереди на обслуживание ферментом. Наблюдаемое явление проще всего объяснить именно через формирование субстратом и ферментом некоего промежуточного комплекса, что и сделал в 1902 году французский химик В. Анри.
В 1913 году двое немецких биохимиков, Леонор Михаэлис и М.Л. Ментен, приняли это предположение и подвергли его математическому анализу наподобие того, что используется химиками при расчете скоростей обычных реакций. В итоге у них получилось уравнение, описывающее изменение скорости реакции в зависимости от концентрации субстрата, — и математическая модель в точности совпадала с экспериментальными данными. В ходе наблюдений за конкретными реакциями ученые сумели с помощью своих уравнений рассчитать силу, привязывающую определенный субстрат к соответствующему ферменту. С тех пор для подобных целей всегда используются уравнения Михаэлиса— Ментена.
Тот факт, что на основе некоторого допущения можно разработать математическую модель, результаты которой в точности соответствуют экспериментальным данным, конечно, не является однозначным доказательством истинности этих допущений. Но некоторое свидетельство в их пользу все же представляет.
Еще одно подтверждение пришло совсем с другой стороны. Предположим, что строение некоего субстрата очень похоже на строение другого вещества. Что произойдет, если это вещество добавить в раствор фермента вместо нужного субстрата? Так сделал в 1930 году биохимик Дж.Г. Кастел, работая с ферментом, субстратом для которого является янтарная кислота (рис. 33).
По структурной формуле янтарная кислота очень близка к другому веществу, малоновой кислоте (рис. 33). Если в раствор фермента добавить вместо янтарной малоновую кислоту, реакции не происходит. У малоновой кислоты отсутствует одна группа СН2, и этого достаточно, чтобы фермент безошибочно различал их.
Но нельзя сказать, что на добавление малоновой кислоты фермент не реагирует вообще никак. Если сначала добавить в него малоновую кислоту, а потом уже в смесь малоновой кислоты и фермента добавить янтарную кислоту, то реакции опять-таки не будет. Малоновая кислота «отравляет» фермент, или, если выражаться рациональнее, подавляет его действие.
Напрашивается вывод, что молекула малоновой кислоты достаточно похожа на молекулу янтарной, чтобы занять ее место на активной поверхности фермента, но недостаточно похожа, чтобы реакция стала осуществляться дальше. Связь малоновой кислоты с ферментом оказывается достаточно сильной, чтобы не распадаться. Это как если в замочную скважину вставить несоответствующий ключ и сломать его внутри. Теперь дверь нельзя открыть ни этим ключом, ни соответствующим. На самом деле механизм улавливания ферментом конкретного субстрата так и именуется — «замочный механизм».
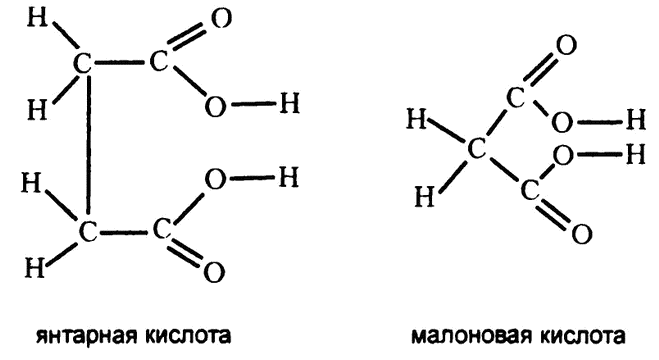
Рис. 33. Молекулы-конкуренты
Малоновая кислота — ингибитор, угнетающее вещество, — конкурирует с «законной» янтарной кислотой за место на рабочей поверхности фермента, поэтому весь процесс носит название «конкурентное ингибирование». С 1930 года изучено уже множество случаев конкурентного ингибирования. Были внесены изменения даже в уравнение Михаэлиса—Ментена для расчета формирования соединений фермента с ингибитором, а не только с субстратом, и снова математические расчеты совпали с экспериментальными данными.
Конкурентное ингибирование может быть методом управления скоростью реакций, катализируемых ферментами в тех случаях, когда по какой-либо причине невозможно физически удалить сам фермент или, наоборот, интенсифицировать его производство. Существует ряд групп важных для организма веществ, сходных по своему строению. Аминокислоты валин, лейцин и изолейцин — похожи. Сахара глюкоза и галактоза — похожи; и так далее. Отношения конкурентного ингибирования между ними практически неизбежны. Похоже, что присутствие в клетке этих веществ в различных соотношениях постоянно производит конкурентное ингибирование тех или иных ферментов в зависимости от их относительной концентрации, таким образом оказывая тонкое влияние на биохимию клетки, изменяя ее в нужную сторону или, наоборот, поддерживая ее в правильном состоянии. Получается своего рода автопилот на молекулярном уровне.
Гораздо более впечатляющих результатов можно добиться с помощью намеренного добавления в организм некоторых веществ. Такие вещества могут иметь лабораторное происхождение и быть полностью чужеродными живой ткани. Такого рода намеренное конкурентное ингибирование позволяет провести различие между организмами даже в том случае, если они тесно переплетены между собой, как, например, паразитирующая бактерия и зараженный организм носителя.
Действие многих ядов обусловливается именно их активным вмешательством в деятельность множества ферментов. Например, такой яд, как дихлорид ртути (сулема), может убить вообще все живое — и микробы, и больного.
А вот если использовать конкурентное ингибирование, то можно вывести из строя один, и только один фермент из всех присутствующих в клетке. При правильно подобранной дозировке можно добиться, чтобы фермент бактерии прекратил свою деятельность, а ферменты человека практически не пострадали — частично из-за повышенной чувствительности фермента бактерии, частично благодаря тому, что ингибитор легче проникнет сквозь клеточную мембрану бактерии, чем человека. А может оказаться и так, что один и тот же фермент нужен больше бактерии, чем человеку, по причине разницы в механизмах обмена веществ.
Первым важным примером такого рода стал сульфаниламид (рис. 34), вещество, впервые синтезированное в 1908 году. В 1932 году один немецкий биохимик занимался исследованием различных красителей на предмет того, могут ли они убивать бактерии, не нанося при этом существенного вреда высшим организмам. Одно из исследуемых веществ, под названием «пронтосил», оказалось очень эффективным средством против некоторых видов стрептококков, о чем и было заявлено на весь мир в 1934 году.
Будучи введенным зараженной лабораторной мыши, пронтосил оказывал свое действие, но вот в пробирке бороться с бактериями отказывался. Соответственно, возникло предположение, что реальную пользу оказывает не сам пронтосил, а некое вещество, формируемое организмом на его основе. Французские биохимики разложили молекулу пронтосила и получили, в частности, составляющую, оказавшуюся сульфаниламидом. Вот она-то и оказалась эффективным средством борьбы с бактериями как в живом организме, так и в лабораторной пробирке.
Так было получено первое из целого ряда «чудо-лекарств», которые в течение одного поколения позволили человеку покончить с инфекционными заболеваниями, довлевшими над ним на протяжении всей истории.
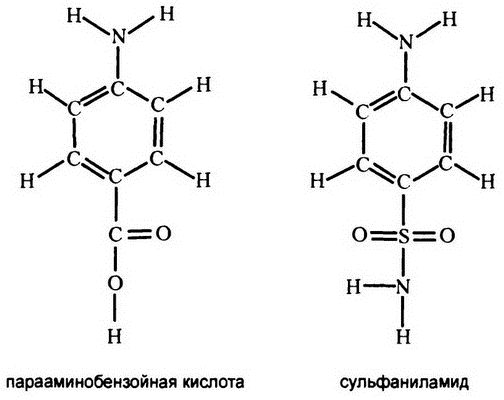
Рис. 34. Молекулы-конкуренты
Как выяснилось, сульфаниламид по структуре своей похож на вещество под названием «парааминобензойная кислота» (рис. 34), которое является важной составляющей всех клеток. Он конкурентно подавляет фермент бактерии, работающий с этим веществом, и, таким образом, убивает бактерию.
С тех пор было разработано много других антибиотиков (пенициллин, стрептомицин и т. п.), каждый из которых работает, очевидно, посредством того или иного конкурентного подавления, хотя какие именно ферменты подавляются в том или ином случае, биохимики пока сказать не могут.
«Замочный механизм», позволяющий ферменту отличать одно вещество от другого, выглядит особенно примечательно в свете «оптически активных соединений». Последний термин необходимо пояснить, чем я сейчас и займусь, — а начну со света.
С давних пор было известно, что луч света, переходя из воздуха в другую прозрачную среду, например воду, — преломляется. Однако в 1670 году датский физик Эразм Бартолин обнаружил, что кристалл исландского шпата (разновидность карбоната кальция, СаСО3) преломляет луч света двумя разными способами одновременно, формируя таким образом два луча из одного. Это явление получило название «двойная рефракция».
Явление двойной рефракции не могла объяснить ни одна теория на протяжении более ста лет. Однако в начале XIX века было установлено, что свет ведет себя так, как если бы он состоял из крошечных волн, и с такой точки зрения вопрос о двойной рефракции достаточно быстро оказался решенным.
В обычном луче света, например исходящем от солнца или любого другого нагретого предмета, световые волны колеблются во всех возможных направлениях. Одни колеблются вверх-вниз, другие — влево-вправо, третьи — где-то между первыми и вторыми… Но упорядоченная атомная структура исландского шпата (напомню — теория атомного строения тоже утвердилась в начале XIX века) оставила световым волнам возможность колебаться только в двух направлениях, под прямым углом друг к другу.
Соответственно, при попадании в кристалл формируются два вида световых лучей — в одном из них волны колеблются только вверх-вниз (к примеру), а в другом — только влево-вправо. У этих двух лучей разные свойства, и они преломляются с разным углом, поэтому попадающий в кристалл луч разделяется надвое.
Луч света, волны которого колеблются только в одной плоскости, называется «поляризованным». Этот довольно неудачный термин ввел французский инженер Этьен Луи Малюс, открывший в 1810 году, что волна света, отраженного от стекла под определенным углом, тоже начинает колебаться только в одной плоскости.
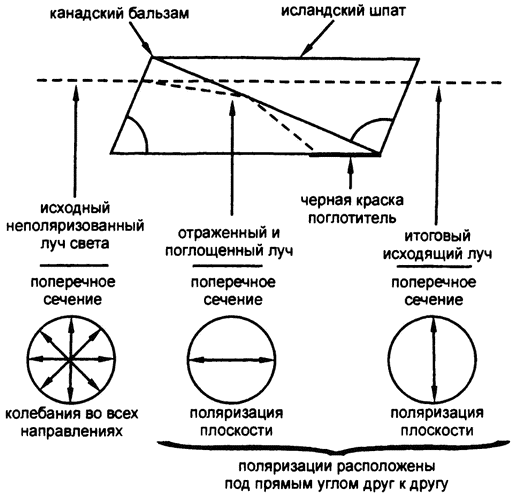
Рис. 35. Призма Николя
Были разработаны методы определения того, в какой именно плоскости колеблется свет конкретного луча; автор метода, лучшего из всех, изобретенного в 1828 году, — британский физик Уильям Николь. Он составил вместе две призмы исландского шпата и склеил их канадским бальзамом таким образом, чтобы один из поляризованных лучей проходил насквозь, а второй — отражался по линии соединения. Получался только один, поляризованный, луч (рис. 35).
Исходящий луч может пройти еще через одну призму Николя, если она ориентирована точно параллельно первой. Если вторую призму Николя начать медленно вращать, то будет видно, что сквозь нее проходит все меньше и меньше поляризованного света, до тех пор, пока вторая призма Николя не окажется под прямым углом по отношению к первой — тогда свет не сможет проходить вообще. Устройство с двумя такими призмами называется «поляриметр». Сейчас вместо дорогих и громоздких призм Николя используются пластиковые листы, внутри которых находятся правильным образом ориентированные органические кристаллы. Их называют «поляризационными светофильтрами».
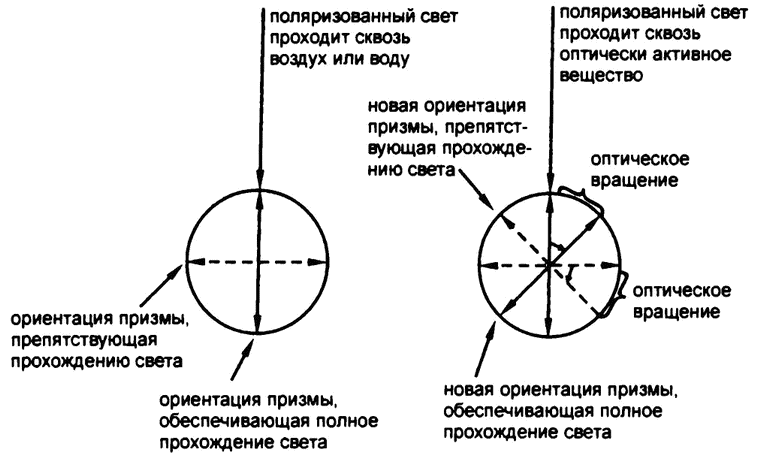
Рис. 36. Оптическая активность
Если луч поляризованного света проходит по пути из одной призмы Николя в другую через воздух или воду, с плоскостью его колебания ничего не происходит. Сквозь вторую, параллельно ориентированную, призму свет проходит без каких-либо существенных изменений.
Однако в 1815 году французский физик Жан Батист Био обнаружил, что если поляризованный свет проходит сквозь кристалл кварца, то плоскость его колебаний при этом изменяется. После того как поляриметр был доработан, оказалось, что свет, пропущенный через кристалл кварца, становится заметно тусклее, проходя через вторую призму Николя, — ее опять приходилось вращать до тех пор, пока она не оказывалась ориентированной параллельно новой плоскости колебаний световой волны, измененной под влиянием кварца. Угол этого изменения можно измерить с достаточной точностью по изменению угла ориентации второй призмы Николя, необходимого для того, чтобы свету вернулась прежняя яркость (рис. 36).
Кроме того, Био показал, что и при прохождении поляризованного света сквозь растворы некоторых органических веществ, например скипидара или камфоры, плоскость его колебаний тоже может изменяться. Именно это свойство вещества и именуется «оптической активностью». Био считал, что причиной ее должна служить какая-то асимметрия в структуре оптически активных веществ, приводящая к искривлению луча света всегда в одном и том же направлении. По его мнению, в «симметричных» веществах плоскость колебаний света тоже искривляется, но одинаково во всех направлениях, так что внешне никаких изменений незаметно.
В 1848 году молодой французский химик Луи Пастер (тогда ему было только двадцать шесть лет, и ему еще предстояло сделать одну из самых блестящих карьер в научном мире) ошеломил ученую общественность тем, что доказал, что так оно и есть — но крайней мере, в отношении кристаллов (рис. 37). На примере кристаллов виннокислого аммония он обнаружил, что кристалл выделяется в двух видах, оба из которых асимметричны и являются точным зеркальным отражением друг друга, так же как руки и ноги человека.

Рис. 37. Двусторонняя асимметрия
Пастер очень осторожно, с помощью микропинцета и увеличительного стекла, разделил кристаллы на две кучки — левосторонние в одну, правосторонние в другую, — и оказалось, что раствор каждой из этих кучек проявляет свойство оптической активности! Если же оба раствора смешать, то в получившейся смеси оптической активности не наблюдается.
Разделение равномерной смеси кристаллов обоих видов надвое по разделительному признаку — это увеличение упорядоченности, а значит — уменьшение энтропии. В нашем примере с игральными картами это равносильно тому, чтобы взять перетасованную колоду и рассортировать на две половины, красные отдельно, черные отдельно. Перед нами, кстати, замечательный пример того, как локальное уменьшение энтропии происходит за счет ее глобального увеличения. Пастеру приходилось напряженно вглядываться в увеличительное стекло и аккуратно брать кристаллики микропинцетом. Понятно, что увеличение его энтропии имело гораздо большее значение, чем произведенное им уменьшение энтропии среди кристаллов.
Так или иначе, Пастер сумел доказать наличие асимметрии только для кристаллов. А в растворах оптически активные вещества представлены в виде молекул. Так где же там может скрываться асимметрия? В самих молекулах, что ли?
Правильный ответ — да! Он был получен в 1874 году, когда двое молодых химиков, голландец Якоб Хендрик Вант-Гофф (двадцать два года) и француз Жозеф Ашиль ле Бель (двадцать семь лет) независимо друг от друга выдвинули новую теорию атома углерода, с помощью которой удалось усовершенствовать систему записи формул Кекуле с учетом некоторых новых фактов.
При изображении структурных формул по методике Кекуле четыре связи, которыми обладает углерод, принято рисовать торчащими во все четыре стороны, что естественно для двухмерного изображения. Однако такой вид не отражает реальности, поскольку если бы все было так, то, например, существовало бы две разновидности метиленхлорида (СН2Cl2), соответствующие двум формулам:
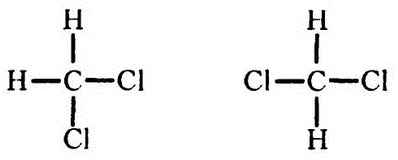
где в одном случае атомы хлора находятся по соседству, а во втором — нет. Однако на практике до сих пор обнаружен пока только один метиленхлорид.
Теория Вант-Гоффа—Беля гласит, что четыре связи углерода направлены в двух перпендикулярных друг другу плоскостях (рис. 38). Изобразить это в двух измерениях сложно, проще всего представить себе равностороннюю пирамиду, вершина которой смотрит в небо. Направления четырех связей — из центра к углам пирамиды, соответственно, угол между всеми связями — один и тот же, около 109°. Атом углерода можно переворачивать как угодно любой из связей вверх — общая картина от этого не изменится.
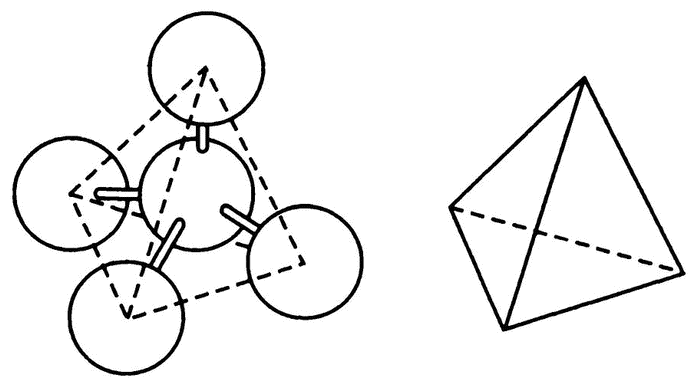
Рис. 38. Атом углерода — тетраэдр
Каждая из этих связей находится на равном расстоянии от остальных, так что если метиленхлорид составлен именно таким образом, то совершенно не имеет значения, на каких именно связях закреплены два атома хлора. Все связи одинаковы. Вы можете сами проверить это на модели: воткните в пластилиновый или восковой шарик четыре палочки в виде описанного мной треножника. Пусть две палочки будут белыми (допустим, это связи с водородом), а две — зелеными (связи с хлором). Попробуйте поменять их между собой, и вы убедитесь, что аналогичного эффекта можно добиться и простым переворачиванием треножника. Соответственно, согласно теории Вант-Гоффа—Беля, метиленхлорид имеет только одно возможное представление — и это согласуется с практическими данными.
Однако, если к каждой из четырех связей углерода прикрепить по разной группе, результат будет уже другим. В этом вы тоже можете убедиться, поэкспериментировав с пластилиновым шариком и четырьмя палочками. Получится, что в таком случае существуют две возможные комбинации, каждая из которых является зеркальным отражением второй. Вы увидите, что, сколько ни крути модель, перевести одно из них в другое не получится. По системе Кекуле эти зеркальные варианты можно отобразить, как показано на рис. 39, и перевести одну формулу в другую невозможно.
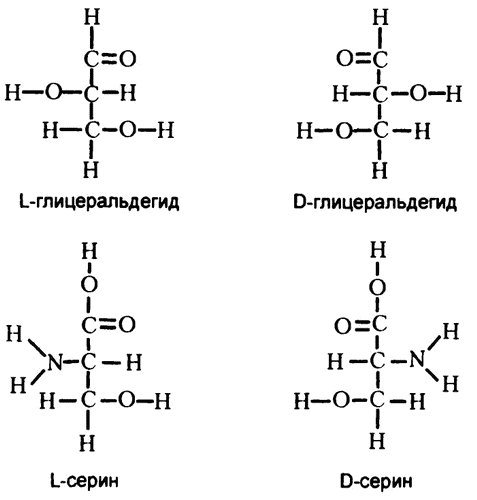
Рис. 39. Левосторонние и правосторонние вещества
Вскоре было установлено, что в каждом случае, когда органическое вещество выказывало оптическую активность, в молекуле обнаруживалась асимметрия, обусловленная наличием как минимум одного «несимметричного углерода» (такого, к которому присоединяется четыре различные группы). При прохождении через вещества одной категории угол плоскости света смещался по часовой стрелке, другой — против часовой. Так при какой же куда?
Казалось, что ответить на этот вопрос невозможно, поэтому в 1891 году немецкий химик Эмиль Фишер вынес по этому поводу произвольное решение. Он выписал на бумаге два варианта строения вещества под названием глицеральдегид (который можно рассматривать как крайне простую молекулу сахара), и пометил один из них как D-глицеральдегид («правосторонний»), а второй — как L-глицеральдегид («левосторонний»). Все остальные оптически активные вещества теперь осталось рассмотреть по аналогии с этими двумя и отнести либо к «левосторонней», либо к «правосторонней» группам. Совсем недавно оказалось, что Фишер действительно был прав. Распределенные им по группам вещества действительно имеют именно то строение, которое он предполагал.
Когда в лабораторных условиях создается какое-либо из веществ, имеющих асимметричную структуру, оба зеркальных варианта формируются в равных количествах, поскольку такова ситуация максимального беспорядка, а следовательно, максимальной энтропии. Для того чтобы вещества одной категории получалось больше, чем другой, необходимо идти на специальные ухищрения, например использовать в качестве сырья другое оптически активное вещество.
Такие вещества добываются из животных и растительных источников. На самом деле все присутствующие в живой ткани вещества, имеющие в своем составе асимметричный атом углерода, представляют собой либо одну, либо вторую оптически активную форму. В этом отношении живая ткань являет собой поразительный пример механизма уменьшения энтропии — как и в отношении создания специфических белков. Естественно, все это делается за счет глобального ее увеличения.
Выяснилось, что все природные сахара — «правосторонние», а все природные аминокислоты (в том числе приведенный для иллюстрации на рис. 39 серии) — «левосторонние». То есть в общем случае организм не может использовать ни левосторонние сахара, ни правосторонние аминокислоты, поэтому их также называют «неестественными» сахарами и аминокислотами. Однако в небольшом количестве в природе они все же встречаются. В состав стрептомицина входит левосторонний сахар, а в состав некоторых антибиотиков — правосторонние аминокислоты. Жизненно необходимое человеку вещество «аскорбиновая кислота» имеет отношение к «неестественным» левосторонним сахарам.
Тот факт, что все природные аминокислоты — левосторонние, крайне важен для пептидной структуры. Если задаться целью создать модель пептидной цепочки с использованием тетраэдрального представления углеродных связей, то окажется, что радикалы звеньев-аминокислот в цепочку направлены по очереди в разные стороны. Тогда вдоль цепочки получается достаточно места для всех групп атомов. Если бы все аминокислоты были правосторонними, ситуация была бы той же.
Однако, если бы часть аминокислот были левосторонними, а часть — правосторонними, то неизбежными стали бы случаи соседства в цепочке левосторонней и правосторонней аминокислот. Их радикалы оказались бы на одной стороне цепочки, и места для обоих просто не хватило бы. Именно поэтому все аминокислоты в белке должны быть либо левосторонними, либо правосторонними. Тот факт, что они оказались левосторонними, видимо, является результатом случая, произошедшего давным-давно в доисторические времена, когда появилась на свет первая белковая молекула.
Молекулы ферментов (тоже сами по себе оптически активные и асимметричные, благодаря наличию в их составе аминокислот), разумеется, отличают оптически активный субстрат от его зеркального отображения, так же как мы не надеваем на правую ногу левый ботинок. Реагируя с одним вариантом зеркального отображения и не реагируя с другим, ферменты поддерживают асимметрию в организме.
Для того чтобы ферменты могли проводить подобное различие между зеркальными вариантами вещества, субстрат должен крепиться к молекуле фермента как минимум в трех точках, и это — еще одно косвенное свидетельство в пользу образования промежуточного комплекса «субстрат-фермент».
Кстати, в 1949 году гипотеза о промежуточном образовании получила окончательное подтверждение — британский биохимик Бриттон Ченс смог непосредственно обнаружить наличие этого комплекса с помощью очень тонкого теста на светопоглощение.
До сих пор я рассуждал так, как будто в состав белков не входит ничего, кроме аминокислот. Да, в отношении большей части белков это так и есть. Однако при распаде многих белков, помимо аминокислот, обнаруживались и отличные от них вещества. Такие белки называют «составными», и самым известным из них является гемоглобин, придающий крови характерный красный цвет.
В 60-х годах XIX века немецкий биохимик Феликс Хоппесейлер сумел показать, что частью структуры гемоглобина является вещество «гем», являющееся не аминокислотой, а сложной группой атомов, содержащей один атом железа. Позже были открыты и ферменты, важную составную часть которых тоже представляет собой гем.
Гем называют «простетической группой» (от греческого слова, означающего «добавление», от которого происходит и слово «протез»), и он необходим для функционирования тех белков, частью которых является. Гем в составе гемоглобина и различных гемсодержащих ферментов — это одно и то же вещество, но у каждого белка имеется своя отдельная функция, определяемая аминокислотной составляющей. Тут можно провести аналогию с режущей кромкой инструментов — топора, серпа, стамески… Режущая кромка одинакова, но предназначение инструмента зависит от его формы в целом.
Гем прочно прикреплен к остальной части белковой молекулы, и удалить его можно, только разложив саму молекулу. Во всех прочих случаях первыми отваливаются аминокислоты. К примеру, некоторые ферменты в процессе очистки диализом теряют каталитические свойства. Причина этого кроется не в денатурации, поскольку, если ту же самую воду, которой промывали мембранный мешочек, потом долить в раствор фермента, он снова обретает свои каталитические свойства. Явно какая-то важная часть фермента в этом случае утрачивается, а потом, вместе с водой, возвращается обратно.
Впервые это продемонстрировали в 1904 году английские химики Артур Харден и У.Дж. Янг, проведя диализ фермента зимазы, полученного из дрожжей. Ту часть его, которая была удалена в процессе диализа, ученые назвали «козимазои». Если она так легко удаляется и так же легко возвращается на место, решили химики, значит, удерживающие ее в составе молекулы связи должны быть очень уж слабыми. Кроме того, молекула козимазы должна иметь достаточно малый размер, чтобы проникнуть сквозь мембрану. И еще — выяснилось, что кипячение не разрушает структуру козимазы, следовательно, это не белок.
Потом и у других белков обнаружилось свойство терять небольшие частицы и утрачивать при этом свои характерные способности; тогда эти «небольшие частицы» получили общее имя — «коферменты».
Работа по установлению химической природы коферментов ведется с 20-х годов XX века по сей день. В составе большей части из них был обнаружен атом фосфора, а многие содержали такие сочетания атомов, каких в организме не обнаруживалось более нигде. Например, в составе козимазы было найдено кольцо из пяти атомов углерода и одного атома азота, как в соединении, имеющем название «пиридин». Сам по себе пиридин в живых тканях не встречается, как и пиридиновое атомное кольцо, за исключением нескольких коферментов. То же самое можно сказать еще о ряде групп атомов, имеющихся в других коферментах.
Ферменты присутствуют в организме лишь в небольших, достаточных для осуществления катализа количествах, так что и коферменты, особенно содержащие такие редкие соединения атомов, присутствуют в крайне малом количестве.
Этот факт имеет значение в свете другой линии исследования, которая проходила независимо от той, о которой я только что рассказывал. На протяжении всей своей истории человек периодически отмечал существование определенной связи между питанием и некоторыми заболеваниями. К примеру, в 1753 году шотландский врач по имени Джеймс Линд предположил, что наличие в рационе цитрусовых и свежих овощей позволяет предотвратить цингу. Эта болезнь, выражавшаяся в кровоточении десен и общем ослаблении организма, была настоящим бичом моряков, которым приходилось иногда по нескольку месяцев сидеть на диете из солонины и сухарей. Несколько десятков лет спустя командование британского флота решило прислушаться к совету врача и включить в диету моряков лимоны — цинга просто испарилась с британских кораблей.
В 1896 году голландский врач Кристиан Эйкман, работая в Восточной Индии, сумел воспроизвести у домашней птицы симптомы распространенного среди местного населения заболевания берибери только за счет кормления их шлифованным рисом. Вылечить болезнь удалось так же просто — добавив в пищу птицы рис нешлифованный.
Очевидно, в рисовой шелухе, которая при шлифовке удаляется, содержится небольшое количество некоторого вещества, необходимого для здоровой жизни. В течение всего следующего десятилетия биохимики обнаруживали подобные вещества и в других пищевых продуктах. Выяснилось, что в состав вещества, содержащегося в рисовой шелухе, входит аминогруппа, и Казимеж Функ, биохимик польского происхождения, исходя из предположения, что и в других подобных веществах есть аминогруппа, назвал их все скопом «витаминами» («жизнь + амины»). Потом оказалось, что аминогруппа — совсем не обязательный элемент витаминов, но название к тому времени уже прижилось.
В течение 30-х годов XX века, да и позже, ученые выделяли все новые витамины и выясняли их строение. И чем дальше, тем больше становилось очевидным, что в витаминах содержатся те же самые редкие группы атомов, что и в коферментах. Выводы, мне кажется, напрашиваются сами.
Организм живого существа, например человека, не может самостоятельно производить некоторые «необычные» сочетания атомов, потому что для этого у него отсутствует соответствующий химический механизм. А вот растения способны такие вещества вырабатывать, и травоядные животные получают их вместе с пищей. Так эти вещества, в небольшом количестве, попадают и в организм человека, но, как я уже говорил, небольшого количества оказывается вполне достаточно, так как и коферментов в организме очень мало.
При нормальном питании в организм человека поступают все необходимые вещества. Однако если диету искусственно ограничить, например питаться лишь тем, что может пролежать без холодильника весь долгий путь морского путешествия, или переусердствовать при очистке зерна, то при этом из пищи пропадает необходимая доза витаминов. А без них организм не может формировать соответствующие коферменты. Когда доходит до этой стадии, ферменты, в состав которых эти коферменты должны были бы входить, становятся недееспособными, внутриклеточные химические механизмы начинают буксовать, и человек заболевает, ему становится все хуже, и если положение не исправить, то в конце концов он умрет.
Эти неаминокислотные составляющие белков не являются по природе своей чисто органическими веществами. Я уже сказал, что для жизнедеятельности человека необходимо железо, поскольку оно входит в состав гемоглобина и гемсодержащих ферментов. Более того, оно необходимо в приличных количествах, поскольку гемоглобин составляет значительную часть крови.
Однако некоторые ферменты присутствуют в организме в гораздо меньшем количестве, нежели гемоглобин, и содержат уникальные, присущие в организме только им элементы — соответственно, в целом содержание этих элементов в организме не то что просто мало, а ничтожно мало. Например, фермент карбонангидраза содержит в своем составе цинк. В состав других ферментов входят магний, медь, кобальт или молибден. Для того чтобы обеспечивать потребности человека в этих веществах, они должны присутствовать в рационе в минимальном объеме, но полное их отсутствие приведет к нарушению работоспособности важнейших ферментов и, соответственно, к болезни и смерти.
Более 800 000 книг и аудиокниг! 📚
Получи 2 месяца Литрес Подписки в подарок и наслаждайся неограниченным чтением
ПОЛУЧИТЬ ПОДАРОКЧитайте также
Глава 21 Микробы и ферменты
Глава 21 Микробы и ферменты Около 100 лет тому назад в 1907 г. Эдвард Бюхнер (не путать с Эрнестом Бюхнером, изобретателем воронки Бюхнера) получил Нобелевскую премию за открытие ферментации бесклеточными экстрактами из дрожжей. Растерев дрожжевые клетки с кварцевым песком и
Ферменты служат клетке
Ферменты служат клетке В живых клетках происходят многие химические реакции, воспроизвести которые в лаборатории оказалось возможным лишь при создании специфических условий. Одни из них протекают при высоких температурах, другие требуют повышенного давления. Как же
ГЛАВА XV.
ГЛАВА XV. Обработка земли под яровые хлеба.Обработку земли под яровые хлеба я начинаю тотчас после уборки озими . Только при соблюдении этого условия можно рассчитывать на самый обильный урожай.Поля, поросшие сорными травами и покрытые густым жнивьем, я вспахиваю
Т–и В–лимфоциты работают сообща, кооперативно.
Т–и В–лимфоциты работают сообща, кооперативно. — Как же кооперируют Т–и В–лимфоциты? Что значит их взаимодействие? Они что–то друг другу передают? Эти события относятся, наверное, к молекулярной биологии. Известны ли они? — Да, в иммунологии несколько
Глава I
Глава I Восемь лет назад я написал небольшую книгу «Аксиомы биологии»[1], в конце которой высказал предположение, что возможно создание общей теории эволюции последовательно реплицирующихся систем. Завершил я книгу словами: «Под эту категорию попадают не только объекты
Глава 3. Ферменты. Механизм действия ферментов
Глава 3. Ферменты. Механизм действия ферментов Ферментами или энзимами называют специфические белки, входящие в состав всех клеток и тканей живых организмов и выполняющие роль биологических катализаторов.Общие свойства ферментов и неорганических катализаторов:1. Не
Ферменты плазмы крови
Ферменты плазмы крови По происхождению ферменты плазмы крови можно подразделить на 3 группы.1. Собственные ферменты плазмы крови (секреторные). Они образуются в печени, но проявляют своё действие в крови. К ним относятся ферменты свертывающей системы крови – протромбин,
Глава 3
Глава 3 Описание метода лечебной кинологии и результатов исследования В предыдущих главах описывались теоретические аспекты метода лечебной кинологии. Постараемся схематически описать практический аспект методологии. Как уже отмечалось, данный метод находится на
Глава 3
Глава 3 Описание метода лечебной кинологии и результатов исследования В предыдущих главах описывались теоретические аспекты метода лечебной кинологии. Постараемся схематически описать практический аспект методологии. Как уже отмечалось, данный метод находится на
Глава 8 Он и она
Глава 8 Он и она Равенство не означает одинаковость Мужчины и женщины ведут себя по-разному во многих ситуациях. В этом нет сомнения. Основной вопрос, который вызывает споры уже много тысяч лет: предопределены ли различия в их поведении природой или же они – результат
Ферменты, открытые дважды
Ферменты, открытые дважды История науки изобилует множеством драматических моментов, когда блестящие открытия, очевидные для потомков, совершенно не воспринимались современниками. Так, например, было с открытием нуклеиновой кислоты молодым швейцарским врачом Иоганном