Глава 6 Биологическая химия
Формирование биологической химии как самостоятельной дисциплины в системе биологических наук было длительным и сложным процессом. Современная биологическая химия сформировалась на рубеже XIX и XX вв. Основными факторами ее формирования было развитие химии важнейших природных соединений — жиров, углеводов и особенно белков, первые успехи энзимологии, разработка основных положений о многоступенчатости обмена веществ и роли ферментов в этих процессах. Главной целью биологической химии стало изучение методами химии не суммарных процессов обмена веществ, а превращений в организме каждого отдельного соединения и разработка представлений о всех деталях обменных процессов в их совокупности.
В первой половине XX в. были сделаны кардинальные открытия, позволившие построить общую схему обмена веществ, установить природу ферментов и исследовать их важнейшие свойства, значительно расширить знания о других биологически активных соединениях. В 40-50-е годы быстрыми шагами пошло развитие и усовершенствование биохимических методов исследования. В настоящее время биохимия определяется как «наука, изучающая состав организмов, структуру, свойства и локализацию обнаруживаемых в них соединений, пути и закономерности их образования, последовательность и механизм превращений, а также их биологическую и физиологическую роль»[52].
Биохимические исследования охватывают очень широкий круг проблем: по существу сейчас нет ни одной отрасли теоретической или прикладной биологии и медицины, которая не была бы теснейшим образом связана с биохимией.
Применение органического анализа для изучения химических процессов в организме.
Использование методов химии в исследованиях жизнедеятельности биологических объектов началось вместе с рождением новой химии.
Уже А. Лавуазье, изучая в 1776 г. процесс горения, одновременно проследил химические изменения, которые претерпевает воздух в организме животных. Создание теории горения позволило А. Лавуазье и П.С. Лапласу впервые коснуться проблемы энергетики обмена веществ организмов. 18 июня 1789 г. они доложили Парижской академии об экспериментах по определению теплового баланса химических и физиологических процессов.
Метод сжигания А. Лавуазье как первый метод органического анализа позволил в начале XIX в. приступить к исследованиям химического состава различных продуктов животного и растительного происхождения. Среди первых веществ, подвергнутых достоверному элементарному анализу французскими учеными Ж.Л. Гей-Люссаком и Л.Ж. Тепаром в 1810 г., были яичный белок, фибрин крови и казеин. Естественно, что наибольшее внимание химиков привлекали основные компоненты организмов — жиры, углеводы и белки. Интерес к первым двум группам веществ стимулировался также потребностями развивающихся сахарных, мыловаренных и свечных производств. Гей-Люссак установил, что сахара являются производными углерода и воды. Вскоре другой французский химик, М. Шеврель, заложил основы химии жиров (1823). В 1933 г. Гей-Люссак установил обязательное присутствие азота в белках растений.
В 30-40-е годы XIX в. голландский врач и химик Г.Я. Мульдер предпринял первую попытку расшифровать химическое строение белковых веществ. Созданная им «теория протеина» была с энтузиазмом встречена физиологами, так как позволяла представить не только конституцию белковых веществ и их производных, но и приблизительную картину их образования и распада, а следовательно, химизм основных процессов обмена веществ. Например, процессы, происходящие в организме при дыхании, после А. Лавуазье описываемые суммарными результатами, стало возможным представить в виде химической реакции образования окислов протеина. Однако вскоре работами Ю. Либиха и его ученика, молодого русского химика Н. Лясковского, было показано, что предложенные Мульдером формулы не соответствуют данным анализа, и теория протеина, несмотря на ее поддержку Я. Берцелиусом, была оставлена.
Уже в первой половине XIX в. возникла тенденция использовать методы химии и физики для изучения не только состава тел, но и протекающих в них физиологических процессов. К этому побуждали интересы медицины и сельскохозяйственной практики. После работ Г. Деви (1813) и Ю. Либиха (1840, 1842) все большее число ученых стало склоняться к мысли о познаваемости жизненных процессов на основе законов химии и физики. Наиболее прозорливые физиологи подчеркивали необходимость изучения химической сущности таких важнейших процессов, как пищеварение и дыхание. При этом надежды, связываемые с физиолого-химическими исследованиями, даже несколько преувеличивались. Так, весьма распространенным было мнение, что химические процессы в организме абсолютно идентичны химическим процессам вне его. В 1875 г. К. Бернар писал: «Дыхательные явления принимались за тип чисто химических»[53]. Подобная переоценка, так же как и распространение виталистических взглядов, определялась многими причинами. Одной из них были недостаточные знания о катализе и, в частности, о биологических катализаторах — ферментах, хотя ферментативное (амилазное) действие было открыто уже в 1814 г. петербургским академиком К. Кирхгофом. Существование ферментов стало бесспорным после работ А. Пайена и Ж. Персо (1833), Ж. Форе (1835), Т. Шванна (1856) и А. Корвизара (1856), выделивших амилазу, синигриназу, пепсин и трипсин.
Но дальше нескольких робких и малоудачных попыток общего химического обоснования важнейших жизненных процессов в 40-60-х годах XIX в. биохимия не пошла.
Первые исследования окислительных процессов.
Наиболее результативными оказались исследования процессов биологического окисления. Повышенный интерес к этим процессам объяснялся следующими причинами. Во-первых, успехи химии в середине XIX в. способствовали выяснению роли в этих процессах кислорода. Во-вторых, в то время полагали, что исследование процессов окисления в организме позволит помимо основной проблемы решить весьма важные для физиологии вопросы о субстрате и месте окислительных процессов в организме.
В 30-х годах XIX в. А.М. Филомафитский и И. Мюллер уже считали ошибочным, что акт дыхания ограничивается пределами легких, полагая, что образование углекислоты может происходить в тканях или крови. Какие именно вещества должны были при этом окисляться, оставалось неясным.
Теория горения и дыхания А. Лавуазье создала предпосылки для исследования сущности окислительных процессов, в том числе биологических. Первую обобщающую теорию окислительных процессов развил швейцарский химик X.Ф. Шенбайн (1845). Его теория, основанная на собственных экспериментах и подкрепленная анализом известных до него фактов, имела целью объяснить сущность окислительных процессов вообще; Шенбайн использовал ее для объяснения окислительных реакций в организме. В его теории биологическое окисление впервые рассматривалось как каталитический процесс. Шенбайн выдвинул представление об активировании кислорода как обязательном и основном условии осуществления окислительных процессов. Создатель перекисной теории биологического окисления А.Н. Бах, отдавая должное работам швейцарского химика, писал: «Потребовалось более полустолетия на более или менее методическую разработку того поля, на котором Шенбайн расставил столько вех»[54].
После работ Шенбайна биологическое окисление, как и окисление вообще, стали трактовать как реакцию присоединения активированного кислорода к окисляющимся телам. Немецкий химик Ф. Гоппе-Зейлер (1878) впервые развил представления об окислении атомарным кислородом (с разрывом молекулы) и указал на важную роль воды в этом процессе, а немецкий физиолог М. Траубе (1882) высказал предположение, что активирование кислорода не обязательно сопровождается разрывом молекулы кислорода, а скорее заключается в образовании перекиси водорода.

Алексей Николаевич Бах. 1857–1946.
Важным этапом в создании представлений о механизмах биологического окисления была перекисная теория биологического окисления, разработанная А.Н. Бахом (1897; Ленинская премия, 1927). Предполагая промежуточное образование перекисей органических соединений или водорода в качестве активированной формы кислорода, эта теория была по существу первой подлинно биохимической концепцией, основанной на представлениях о сложной организации обменных процессов; в начале XX в. она признавалась единственным достоверным объяснением сущности окислительных процессов. По А.Н. Баху, окисление в организме происходит следующим образом:
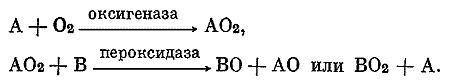
Предположение, что биологическое окисление — это процесс, осуществляющийся в результате двух последовательно скоординированных ферментативных процессов, открыло новые пути в толковании химических преобразований, осуществляющихся в организме.
Утверждение выдвинутых еще Шенбайном представлений о каталитической природе окислительных процессов в организме было одним из важнейших результатов теоретической разработки проблемы биологического окисления. До 80-х годов XIX в. катализаторами биологического окисления считали металлы, или специальные «ферменты», природа и свойства которых оставались неизвестными. Открытие оксидазного действия и выделение первых препаратов биологических катализаторов окисления японским химиком X. Йосидой (1833) и французским физиологом Г. Бертраном (1895) позволило приступить к изучению природы и свойств оксидаз, которые впоследствии были отнесены к классу истинных ферментов.
Параллельное развитие теоретических исследований биологического окисления и оксидаз привело к первым попыткам создания теории оксидазного действия. Это были по существу первые теории ферментативного действия вообще. Наиболее значительными исследованиями в этой области стали работы Г. Бертрана, разработавшего представления о коферментах.
Другие процессы обмена были исследованы гораздо слабее. В течение почти всего XIX в. биологическая химия (вернее, физиологическая химия) не смогла продвинуться дальше изучения суммарных химических эффектов и изменений, т. е. химического изучения еще не расчлененных на отдельные звенья процессов, происходящих в целых организмах или отдельных органах.
Создание теорий химического строения, жиров, углеводов и белков.
Планомерное изучение важнейших биологических соединений и их превращений стало возможным только после того, как стало известно их химическое строение и были разработаны методы их синтеза.
Во введении к «Органической химии, основанной на синтезе», М. Бертло в 1860 г. писал: «До работ, которые изложены в настоящей книге, никакого систематического исследования не проводилось. Можно сослаться на два примера полного синтеза естественных тел из элементов: на синтез мочевины Вёлером и синтез уксусной кислоты Кольбе. Эти синтезы были очень интересны, но вследствие природы полученных при этом тел они оставались изолированными и неплодотворными. История науки свидетельствует о том, что два упомянутых синтеза не послужили отправным пунктом ни для одного общего метода и не приводили даже к какому-либо другому необычному воспроизведению природных тел»[55].
С именем самого М. Бертло связано решение проблемы строения и синтеза жиров. Начав исследования глицерина и его производных, он установил в 1854 г., что глицерин является многоатомным спиртом. Бертло синтезировал его моно-, ди- и триацетаты. Синтезировав затем ряд аналогичных эфиров, он окончательно установил, что природные жиры представляют собой сложные эфиры жирных кислот.
Гораздо более сложным оказалось исследование строения другой группы природных веществ — сахаров. После того, как Т. Соссюр (1804) выдвинул общую теорию превращения углекислого газа и воды в органические соединения под действием солнечного света, поддержанную Ш. Дюма и Ю. Либихом, наиболее значимым достижением, проливающим свет на процессы синтеза углеводов в растениях, было установление Ж. Буссенго (1858) коэффициента ассимиляции, который определяет соотношение объемов превращающегося СО2 и выделяемого О2 (приблизительно 1:1, по Буссенго). Исходя из этого, А. Бейер (1870) предложил уравнение, описывающее эту реакцию:
СО2 + Н2О ? СН2 + О2.
Продукт реакции — формальдегид стал рассматриваться как возможный промежуточный продукт образования сахаров. В 1861 г. А.М. Бутлеров, прибавляя на холоду известковое молоко к раствору триоксиметилена, получил желтый сироп, дающий реакции на сахара. В 1882 г. О. Лёв путем полимеризации формальдегида получил несбраживаемый сироп состава С6Н12О6, а в 1889 г. — сбраживаемое соединение подобного же строения.
Наибольший успех в исследованиях углеводов выпал на долю немецкого химика Э. Фишера. В 1890 г. он предложил простую номенклатуру углеводов. Используя открытый им фенилгидразин, Фишер разработал метод превращения альдогексоз в кетогексозы, например, глюкозы во фруктозу, позволивший установить различия и тождество конфигураций групп СНОН у различных гексоз.
В продуктах конденсации, полученных по Бутлерову и Лёву, Фишер открыл D, L-фруктозу, для получения которой он разработал новый метод — синтез из формальдегида и глицеринового альдегида в присутствии щелочи. Фишер разработал также ряд других методов синтеза альдогексоз, полностью подтвердивших выдвинутые им представления о строении сахаров (Нобелевская премия, 1902).

Эмиль Фишер. 1852–1919.
После изучения углеводов и исследований соединений пуриновой группы (аденин, гуанин, мочевая кислота, кофеин, теобромин, гипоксантин и др.) Фишер приступил к анализу аминокислот и белков.
К концу XIX в. после работ Либиха выяснилось, что дифференцировать белки по их элементарному составу невозможно и даже ведущие химики не верили в возможность выяснить строение белковой молекулы. Поэтому приходилось ограничиваться анализом продуктов частичного распада белковых веществ.
В 1870 г. Н.Н. Любавин впервые высказал мысль о том, что белки состоят из аминокислот. С 1806 по 1890 г. в составе белков было открыто девять аминокислот. В основе теорий строения белков, предложенных А.Я. Данилевским, П. Шютценберже и особенно А. Косселем (Нобелевская премия, 1910), лежала идея о том, что белки построены в основном из аминокислотных остатков. Эти работы стимулировали поиски новых аминокислот в белках. В результате в течение последнего десятилетия XIX в. было открыто еще четыре аминокислоты.
Пептидная теория строения белка.
До начала XX в. методов определения содержания аминокислот в белках не существовало, химия аминокислот была изучена весьма слабо, а попытки синтеза белковых веществ методологически были совершенно неправильными.
Приступая к систематическому изучению белков, Э. Фишер исходил из представления, что белки построены только из аминокислот, и попытался доказать наиболее вероятное, с его точки зрения, амидное соединение аминокислотных остатков. Он разработал метод разделения смесей аминокислот (так называемый эфирный метод) и использовал его для выделения и идентификации отдельных аминокислот из продуктов кислотного, щелочного или ферментативного разложения белков. После этого он перешел к решению ключевого вопроса о характере связей отдельных аминокислот в молекуле белка. Фишер пошел при этом по новому пути. Вместо применявшегося ранее исключительно аналитического метода исследования он использовал метод синтеза: он добивался получения из аминокислот все более и более сложных соединений и пытался их идентифицировать с продуктами частичного распада белковой молекулы — пептонами. В 1901 г. Э. Фишер и Э. Фурно сообщили о синтезе первого пептида — глицил-глицина. В статье, положившей начало систематическому изучению синтетических полипептидов, авторы писали: «Чтобы в этой трудной области получить достоверные результаты, прежде всего необходимо найти метод, который позволил бы последовательно соединять друг с другом молекулы различных аминокислот при помощи связей ангидридного типа, при условии, что каждое из полученных промежуточных соединений может быть охарактеризовано»[56].
Разработав в течение 1902–1919 гг. несколько методов синтеза соединений аминокислот, так называемых пептидов и полипептидов различного строения, среди которых был, в частности, октадекапептид, состоящий из 18 аминокислотных остатков, Фишер доказал, что основным типом связи аминокислот в молекуле белка является амидная связь между аминогруппой одной аминокислоты и карбоксилом другой. Эту связь Фишер назвал пептидной. Пептидная теория удачно объяснила многие основные свойства белковых веществ — как химические и физико-химические, так и биологические.
В 1925–1929 гг. А.Р. Кизель опроверг ошибочное представление о том, будто непременной основой всякой протоплазмы является особое тело белковой природы — пластин, с которым связаны ее важнейшие структурные и функциональные особенности. Благодаря исследованиям Кизеля изучение белков стало на правильный путь (формирование современных представлений о строении и функциональной роли белков изложено в главе 23).
Первые успехи в изучении природы биокаталитических реакций. Открытие специфичности действия ферментов.
В последней четверти XIX в. было установлено, что некоторые химические реакции, с большой легкостью протекающие в организме, чрезвычайно трудно осуществить вне его. На ранних этапах развития энзимологии представление об универсальности ферментативных механизмов оспаривалось вследствие того, что исследования ферментов связывались с отдельными технологическими или физиологическими процессами. Было распространено, например, мнение, что функция ферментов в процессах пищеварения ограничивается разложением пищи. Открытие и изучение окислительных ферментов в последней четверти XIX в. привели к представлению о каталитическом характере ферментативных реакций и выявили их широкое распространение в природе.
Доказательство того, что ферментативные реакции являются разновидностью химических реакций, имело принципиальное значение в борьбе с витализмом. В 1890 г. Ч. О’Селливан и Ф. Томпсон провели исследование действия дрожжевой инвертазы чисто количественными методами и пришли к заключению, что инверсия следует классическим химическим закономерностям и представляет собой мономолекулярную реакцию. В 1892 г. Г. Тамман сформулировал положение о том, что «неорганизованные ферменты ускоряют гидролитические реакции так же, как и кислоты»[57].
Впервые на значение возможности осуществления обратимых биокаталитических реакций указал в 1878 г. А.Я. Данилевский, исследуя ресинтез белков из продуктов их распада в присутствии протеолитических ферментов. Открытие синтетических возможностей ферментов было подтверждено работами А.К. Хилла, осуществившего ферментативный синтез мальтозы (1898).
Изучая строение сахаров, Э. Фишер начал в 1894 г. ряд исследований, касающихся действия на них ферментов. Он убедился, что разные субстраты по-разному атакуются ферментами, а последние в свою очередь обладают избирательностью при действии на различные субстраты. В итоге он пришел к открытию специфичности действия ферментов. Это открытие сделало возможным изучение пределов действия ферментов и внесло гораздо большую определенность в характеристики отдельных катализируемых ферментами реакций и самих ферментных систем.
Исследования Фишера развивали уже сложившуюся тенденцию химического подхода к исследованиям ферментов. Его знаменитый афоризм о том, что фермент подходит к субстрату, как ключ к замку, способствовал развитию представлений о стерическом соответствии между ферментом и субстратом. Под влиянием открытий Фишера шло быстрое проникновение достижений химии биологически важных соединений в энзимологию, что привело в итоге к радикальной перестройке всей системы взглядов на методы изучения реакций обмена веществ.
Ученик Фишера Э. Армстронг в 1904 г. установил, что специфичность проявляется и при торможении ферментативной деятельности аналогами субстрата. Его работы так же, как и исследования А. Брауна (1902), легли в основу гипотез о механизме действий ферментов и образовании фермент — субстратных комплексов, что в свою очередь позволило развить представления о кинетике и механизме действия ферментов задолго до выделения их в чистом виде. Так, В. Анри (1903) впервые вывел кинетические уравнения ферментативной реакции, а Л. Михаэлис и М. Ментен (1913) разработали первую кинетическую теорию действия ферментов.
Успехи органического синтеза после создания А.М. Бутлеровым теории химического строения, возникновение стереохимии и совершенствование органического анализа позволили к концу XIX в. развернуть широкие исследования, в результате которых было открыто строение важнейших химических компонентов организма и заложены основы дальнейшего прогресса в изучении обменных процессов.
На рубеже XIX и XX вв. первые успехи в расшифровке биохимических механизмов жизнедеятельности получили противоречивые истолкования. С одной стороны, их совершенно справедливо рассматривали как важное достижение на пути создания более общих представлений о химических механизмах сложных биологических процессов, как закономерную ступень познания сущности жизни и процессов жизнедеятельности. С другой стороны, их или истолковывали в виталистическом духе, как это сделал, например, лидер неовитализма Г. Бунге, или рассматривали как свидетельство торжества механистического подхода к изучению живой природы. Однако, если в XIX в. приверженцы виталистических и механистических концепций пытались найти истину в споре друг с другом, то с начала XX в. решающее значение в определении места химических закономерностей в жизнедеятельности организмов приобрел эксперимент.
Разработка биохимических основ учения о питании. Открытие витаминов.
Наряду с общими проблемами, способствовавшими формированию современной биохимии, на рубеже XIX и XX вв. изучались и некоторые частные вопросы. Зачастую они имели принципиальное значение и получали выход в прикладные области исследования. Наибольшие успехи были достигнуты в изучении биохимии питания.
После работ Ю. Либиха господствовало убеждение, что три категории питательных веществ — пластические (белки), дыхательные (углеводы и жиры) и минеральные (соли) — способны полностью удовлетворить потребности организма. Эта точка зрения получила дальнейшее детальное развитие в работах учеников и последователей Либиха — К. Фойта, У. Этуотера, М. Рубнера и др. Школами Фойта и Этуотера были проведены многочисленные исследования рационов, предложены первые химические и биологические принципы определения питательной ценности различных веществ (в частности, опубликованы таблицы питательной ценности множества продуктов) и разработаны представления о химическом и энергетическом балансах организма. В 1898 г. сотрудник Этуотера Ч. Лэнгуорти сформулировал «закон питания», согласно которому пища служит двум целям: поставке энергии для поддержания температуры тела и работы и снабжению материалом для построения тела. Было подсчитано даже необходимое соотношение белков, жиров и углеводов как источников энергии (1:2,5:1).
Но уже в 80-х годах было показано, что в отличие от животных, питавшихся естественной пищей, подопытные животные, получавшие смесь очищенных белков, жиров, углеводов и солей, погибали. Было высказано предположение, что искусственные питательные смеси неполноценны из-за отсутствия в них некоторых солей, в первую очередь солей редких элементов.
Выяснению этого вопроса была посвящена работа Н.И. Лунина в лаборатории Г. Бунге в Дерптском (Тартуском) университете, опубликованная в 1880 г. Лунин держал мышей на искусственных диетах, составленных из казеина, молочного жира, молочного сахара, солей молока и воды. Все животные через некоторое время погибали. Мыши же, получавшие цельное молоко, развивались нормально. Из этих экспериментов Лунин сделал вывод, что «в молоке, кроме казеина, жира, молочного сахара и солей, должны содержаться еще другие вещества, которые совершенно необходимы для питания»[58].
В 1896 г. к аналогичным выводам пришел голландский врач К. Эйкман, исследовавший заболевание кур, напоминающее заболевание человека и известное в Японии под названием бери-бери. Ему впервые удалось обнаружить, что в оболочках зерен риса содержится какое-то необходимое для поддержания жизни вещество, ибо добавление этих оболочек к пище предотвращало заболевание. Данные Эйкмана были подтверждены наблюдением над заключенными на о-ве Ява.
Работа Эйкмана, продолженная Г. Грейнсом, а также исследования Т. Осборна, Л. Менделя и особенно Ф. Гопкинса[59] привели к окончательному пересмотру представлений Либиха о факторах питания. Гопкинсу удалось точно доказать (1906), что нет животного, которое могло бы жить на пищевой смеси, состоящей из чистых белков, жиров, углеводов и минеральных веществ.
В 1911 г. были опубликованы исследования польского ученого К. Функа, работавшего в биохимической лаборатории Листеровского института в Лондоне. Основываясь на представлениях о «дополнительных» питательных веществах, развиваемых Гопкинсом, Функ направил свои усилия на их поиски. Ему удалось получить из рисовых отрубей бесцветные кристаллические иглы, раствор которых при введении больному полиневритом (бери-бери) вызвал его быстрое выздоровление. В 1912 г. Функ предложил для выделенного им вещества название «витамин», впоследствии распространенное на остальные вещества подобного рода. Он ввел также термин «авитаминоз» для обозначения заболеваний, возникающих при недостатке в пище витаминов.
Выделение и изучение основных биокаталитических систем.
Интенсивное развитие биологической химии в первые десятилетия XX в. было связано в первую очередь с успехами в изучении процессов обмена веществ. Необходимо отметить, что биохимические исследования, предпринимавшиеся для уточнения физиологических процессов, в это время вызывали возражения. Получаемые биохимиками противоречивые результаты (например, при изучении распада белков в процессе пищеварения), не удовлетворяли биологов. Химики же критиковали результаты биохимических исследований из-за отсутствия необходимой для правильных выводов точности определений, поскольку биохимики редко работали с чистыми материалами и убедительным контролем.
Успехи химии жиров, белков и углеводов, разработка новых специальных методов анализа, привлечение методов органической и физической химии для изучения биологических объектов резко изменили это положение. В распоряжении биологии оказались методы, позволившие перейти к количественным исследованиям процессов, протекающих как в целом организме и его органах, так и в тканях и отдельных клетках. В 20-х годах XX в. на первом плане были проблемы энергетики живого организма и превращения веществ в процессе их обмена. Первоначально предполагалось, что расшифровка механизмов дыхания и утилизации организмом продуктов питания автоматически приведет к решению общей проблемы обмена веществ и энергетики живого организма. Поэтому вопросы энергетики и взаимопревращения веществ в организме в работах таких выдающихся биохимиков первой половины XX в., как В.И. Палладин, С.П. Костычев, О. Варбург, Г. Виланд, О. Мейергоф, Г. Эмбден и другие, оказались тесно переплетенными.
Уже с начала XIX в. было известно, что окись углерода, вода и мочевина являются конечными продуктами сложных превращений, которые претерпевают жиры, углеводы и белки в организме животного. Однако прежде, чем можно было наметить хотя бы гипотетический механизм промежуточных ступеней метаболизма, необходимо было идентифицировать хотя бы основные промежуточные продукты. Одной из важнейших предпосылок успешной расшифровки всех этапов промежуточного обмена веществ был прогресс в исследовании биологических катализаторов-ферментов. Исследования С. Сёренсена (1909), показавшего зависимость активности ферментов от величин pH, и Л. Михаэлиса (1914), заложившего основы кинетики ферментативных реакций, позволили перейти к планомерному изучению факторов, влияющих на активность ферментов.
Особо важное значение приобрели попытки выделения и изучения каталитических систем, осуществляющих основные обменные процессы. Первые серьезные успехи в этой области открыли непосредственные пути к изучению промежуточных продуктов обмена углеводов (см. об этом в главе 7). Дальнейшему прогрессу в изучении углеводного обмена и развитию энзимологии способствовало выяснение О. Мейергофом (1918) важного факта, что кофермент, аналогичный козимазе Э. Бухнера, А. Гардена и В. Юнга, необходимый для осуществления спиртового брожения, содержится и в тканях мышц. Он установил также, что бесклеточные экстракты мышц способны превращать углеводы в молочную кислоту. В 1921–1927 гг. немецкий биохимик Г. Эмбден обнаружил, что при распаде гликогена в мышцах за счет неорганического фосфата также образуются гексозофосфорные эфиры. Тем самым было экспериментально подтверждено принципиальное сходство процессов брожения и дыхания, после чего анаэробная фаза распада углеводов стала считаться единой для процессов брожения и превращений углеводов в мышце. Это способствовало возникновению идеи единства химических механизмов обмена веществ и осознанию возможности создания единой схемы распада углеводов.
Важным достижением в этом направлении было открытие факта, что при распаде углеводов в мышце, так же как и при брожении, образуются фосфорилированные трехуглеродные фрагменты. Заслуга в этом отношении принадлежит Г. Эмбдену (1922), обнаружившему среди продуктов распада гексоз фосфоглицериновую и глицерофосфорную кислоты. Это позволило ему и О. Мейергофу (Нобелевская премия, 1922) построить первую обоснованную схему анаэробного распада углеводов, которая впоследствии была значительно расширена и дополнена, в первую очередь Я.О. Парнасом. Эта схема, расшифровывающая реакцию Хардена-Юнга, остается общепризнанной и в настоящее время.
Успехи в изучении коферментов. Доказательство химической общности коферментов и витаминов.
Создание этой схемы и выделение промежуточных продуктов распада глюкозы сопровождалось изучением ферментов, катализирующих отдельные этапы превращений. «Зимаза Бухнера» оказалась комплексом ферментов, часть из которых была двухкомпонентной, и содержала простетическую группу — козимазу.
Одним из наиболее выдающихся достижений биохимии первой половины XX в. было установление шведским химиком Г. Эйлером (20-е годы) того факта, что козимаза представляет собой нуклеотид адениловой кислоты и что ее присутствие необходимо для нормального функционирования многочисленных ферментных систем, осуществляющих окисление промежуточных продуктов обмена веществ. Козимаза, широко распространенная в различных биологических объектах, оказалась идентичной открытому еще в 1918 г. О. Мейергофом фактору окисления гликогена мышечного сока.
С исследованиями Г. Эйлера[60] связано развитие двух новых направлений в энзимологии. Во-первых, выяснение химической природы кофактора процессов дегидрирования позволило приступить на строго химической основе к созданию представлений о механизмах переноса водорода, к изучению кинетики и многих других физико-химических аспектов проблемы действия ферментов. Во-вторых, они создали предпосылки для систематизированных поисков новых ферментов и кофакторов, объединяющихся по новым принципам.
В 1932 г. немецкие биохимики О. Варбург и В. Христиан при исследовании ферментативного окисления глюкозо-6-фосфата обнаружили, что для осуществления этого процесса необходимо присутствие кофактора, отличного от козимазы (или кодегидразы I). Они назвали его кодегидразой II (см. также главу 7).

Отто Варбург. 1883–1970.
В том же 1932 г. школой О. Варбурга и одновременно венгерскими химиками во главе с А. Сцент-Дьёрдьи были начаты исследования так называемых «желтых ферментов», которые привели к открытию и синтезу Р. Куном рибофлавина (Нобелевская премия, 1938). Исследования Р. Куна и П. Каррера[61] показали, что рибофлавин идентичен уже известному тогда витамину В2. За этим открытием последовали другие, которые не только подтвердили важную роль витаминов в обеспечении функционирования различных ферментных систем, но позволили расшифровать биохимический механизм многих заболеваний и открыть новые звенья обмена веществ.

Владимир Иванович Палладин. 1859–1922.
С исследованиями коферментов брожения связана разработка очень важной концепции о механизме окислительных процессов в организме. Как уже отмечалось, после работ А.Н. Баха общепризнанной стала перекисная теория биологического окисления (см. также главу 7). Однако уже в 1912 г. В.И. Палладин (Ленинская премия, 1929) и немецкий химик Г. Виланд независимо друг от друга разработали новую теорию биологического окисления, в основе которой лежало представление о дегидрировании (отнятии водорода). Включение кислорода, по Палладину-Виланду, происходило лишь на последнем этапе и приводило к окислению до воды отнятого от субстрата водорода. Исследования Г. Эйлера, О. Варбурга и других показали, что к числу дегидрогеназ, широко распространенных в природе, а также коферментов и акцепторов водорода относятся и уже известные кодегидразы I и II.
Решение проблемы аэробного дыхания. Открытие цикла трикарбоновых кислот.
В течение долгого времени считали, что брожение и дыхание представляют собой совершенно независимые процессы. Однако уже Э. Пфлюгер во второй половине XIX в. высказывал предположение о тесной взаимосвязи этих процессов. Окончательно представления о единстве брожения и дыхания были разработаны С.П. Костычевым (1910), согласно взглядам которого взаимосвязь между ними могла быть представлена следующей схемой:

Работы Костылева (1907, 1911) доказали, что начальные фазы аэробного дыхания должны быть сопряжены с конечными фазами анаэробного распада углеводов.

Сергей Павлович Костычев. 1877–1931.
Первые шаги к расшифровке механизма аэробного дыхания сделал после работ Костычева в 30-х годах А. Сцент-Дьёрдьи (Нобелевская премия, 1937). Исследуя дыхание измельченных тканей сердечной мышцы голубя, отличающейся особенно высокой скоростью окислительных процессов, он обнаружил, что постепенно снижающаяся интенсивность поглощения кислорода измельченными мышцами (гомогенатами) может быть восстановлена добавлением солей некоторых органических кислот (янтарной, фумаровой, яблочной и щавелевоуксусной). Наоборот, очень близкая по строению малоновая кислота подавляет интенсивность процесса. Почти одновременно шведский химик Т. Тунберг показал, что мышцы содержат особые ферменты — дигидрогеназы янтарной, фумаровой и яблочной кислот. В дальнейшем американский биохимик Г.А. Кребс обнаружил, что «эффект Сцент-Дьёрдьи» может быть получен также при добавлении к гомогенатам солей кетоглутаровой и пировиноградной кислот, а также аминокислот — глутаминовой и аспарагиновой. Эти открытия навели на мысль, что перечисленные кислоты последовательно превращаются одна в другую в процессе разложения наиболее сложной из них. При этом происходит постепенное окисление органического вещества. Так, была выявлена последовательность: лимонная кислота ? цис-аконитовая кислота ? изолимонная кислота ? кетоглутаровая кислота.

Генрих Виланд. 1877–1957.
Подобные схемы полупили должную оценку после выяснения принципа использования Энергии организмом на отдельных этапах окисления (вернее, дегидрирования) промежуточных продуктов. Супругам К. и Г. Кори, исследовавшим процессы распада и синтеза гликогена в мышцах, удалось не только расчленить процесс превращения гликогена в глюкозу на отдельные этапы, но и воспроизвести синтез гликогена из глюкозы in vitro. Глюкоза была последовательно превращена с помощью соответствующих ферментов в глюкозо-6-фосфат, глюкозо-1-фосфат и гликоген. Самым важным в этих исследованиях было выяснение роли АТФ (аденозинтрифосфорной кислоты) как донатора фосфатных групп, а также открытие процесса восстановления АТФ и АДФ (аденозиндифосфата) на последней ступени (Нобелевская премия, 1947).
После того как было выяснено, что фосфорные эфиры являются промежуточными продуктами распада углеводов, Г.А. Кребс предложил в 1937 г. схему довольно сложного цикла превращений органических кислот, объясняющую все основные моменты их постепенного окисления и образования АТФ на узловых этапах этого процесса (Нобелевская премия, 1953). Таким образом, этот цикл, получивший название цикла трикарбоновых кислот, связал процессы поэтапного окисления органических веществ и постепенного выделения энергии в организме. Наиболее важным в цикле Кребса был этап, связывающий процессы аэробного и анаэробного распада углеводов — декарбоксилирование пировиноградной кислоты и перенос ацетильной группы на щавелевоуксусную кислоту, являющуюся конечным продуктом окисления первого компонента цикла — лимонной кислоты. Разработанный Кребсом цикл в дальнейшем был уточнен и подвергся гораздо большей детализации, но его основные звенья сохранили свое значение.
На период 30-40-х годов приходится формирование основных представлений о дыхательной цепи. После открытия Д. Кейлиным цитохромов (1925) возникла реальная возможность построения единой схемы включения кислорода в окислительные процессы. Эта схема предусматривала как активирование водорода, идею которого отстаивали еще В.И. Палладин и Г. Виланд, так и активирование кислорода, после работ А.Н. Баха развиваемое О. Варбургом. Изучение ферментов дыхательной цепи и введение понятия о переносе электронов и окислительно-восстановительном потенциале закончилось в 40-х годах построением схемы дыхательной цепи, лежащей в основе современных представлений.
В передаче водорода (электрона) от молекулы дыхательного субстрата к кислороду воздуха и в активации обоих элементов основную роль играют отдельные ферменты и ферментативные системы. В изучении дегидрогеназ и оксидаз за последние два-три десятилетия получено много новых данных. Например, использование тяжелого изотопа кислорода О18 позволило X. Хайами и его сотрудникам в 50-х годах подтвердить опытным путем достоверность гипотезы А.Н. Баха о возможности в процессе дыхания прямого присоединения кислорода к дыхательному субстрату. При этом было доказано и существование оксидаз, активирующих молекулярный кислород и делающих его способным реагировать с органическими соединениями. Хайами назвал их фенольными оксидазами, а Г. Мезон предложил эту группу ферментов называть трансферазами кислорода.
Не менее успешно шла в последние годы также разработка проблемы энергетики дыхания. В 50-х годах были получены данные, позволившие разработать основы современных представлений по данному вопросу, согласно которым процесс окисления или восстановления можно трактовать как отнятие от дыхательного субстрата и присоединение к нему электрона.
Создание представлений о системе биохимических обменных процессов.
Одним из важных следствий открытия цикла трикарбоновых кислот был поворот от представления о биохимических процессах в клетке как изолированных реакциях к представлениям о системе обменных процессов, связанных между собой во времени и пространстве. Этому переходу содействовали успехи в изучении промежуточного обмена азотистых соединений и обмена липидов.
Кардинальным открытием в области промежуточного обмена азотистых соединений была расшифровка реакций переаминирования L-амино- и ?-кетокислот в организме. Переаминирование и осуществляющие его ферментные системы были открыты в 1937 г. советским биохимиком А.Е. Браунштейном, что позволило связать воедино систему превращений отдельных аминокислот с циклом трикарбоновых кислот. Считалось, что основные элементы цикла — пировиноградная, ?-кетоглутаровая и щавелевоуксусная кислоты — участвуют в реакциях переаминирования с образованием важнейших аминокислот — аланина, глутаминовой и аспарагиновой — и что дальнейшие реакции переаминирования с соответствующими кетокислотами приводят к образованию всего разнообразия аминокислот. Эти предположения получили подтверждение в исследованиях синтеза в организме животных так называемых незаменимых аминокислот (углеродный скелет которых не может быть синтезирован в животном организме). Введение соответствующих кетопроизводных животным, лишенным незаменимых аминокислот, предотвращало их гибель вследствие образования в организме соответствующих аминокислот. Эти работы содействовали развитию представлений об обмене азота в организме (А. Виртанен, Нобелевская премия, 1945; и др.).
Другим важным доказательством правильности упомянутых предположений послужили работы американского химика Р. Шонхеймера с использованием изотопа азота N15. Он показал, что после введения в организм глицина-N15 или лейцина-N15 в большинстве аминокислот организма быстро обнаруживается N15. Таким образом, цикл трикарбоновых кислот оказался как бы связующим звеном между обменом углеводов и азотных соединений.
Созданию представления о целостной и одновременно динамической обменной системе клетки содействовали и исследования обмена липидов, в первую очередь представления о «метаболическом котле» Р. Шонхеймера, с работ которого началось широкое использование изотопов в биохимических исследованиях.
Существование природных жирных кислот с весьма длинной углеродной цепью представляло серьезную проблему при изучении их распада и синтеза в организме. В начале XX в. немецкий физиолог Ф. Кнооп заложил основу теории распада липидов в организме, получившую название теории ?-окисления. При скармливании собакам фенилзамещенных жирных кислот с углеродной цепью различной длины он обнаружил, что окислению в организме с образованием двухуглеродного фрагмента подвергается СН2-группа жирной кислоты, находящаяся в ?-положении по отношению к карбоксилу. Все природные жирные кислоты обладают четным числом углеродных атомов в цепи. В дальнейшем теория ?-окисления была развита в работах Г. Эмбдена и А. Маркса (1908). Подтверждение ее было получено также при исследовании образования так называемых «ацетоновых тел» у диабетиков — ?-оксимасляной и ацетоуксусной кислот и ацетона.
Гипотеза о синтезе жирных кислот была также основана на представлении, что главным строительным материалом для них служат двухуглеродные фрагменты. Еще в конце XIX в. М. Ненцкий высказал предположение, что основной реакцией образования жирных кислот в организме служит альдольная конденсация ацетальдегидных единиц, следующая за восстановлением гидроксильных групп и окислением концевой альдегидной группы.
В 20-30-х годах получила распространение гипотеза И. Смидли, согласно которой исходными компонентами синтеза жирных кислот являются пировиноградная кислота и ацетальдегид, а затем происходит надстраивание двухуглеродных единиц. Однако после открытия коэнзима А.Ф. Линеном (1951)[62] гипотеза Смидли была отброшена.
Прежде чем вопрос о деталях синтеза жирных кислот приблизился к разрешению, было сделано важное открытие, резко изменившее представления биохимиков и физиологов о принципах работы обменного аппарата. Когда в 1935 г. Р. Шонхеймер начал исследования процессов обмена веществ с использованием меченых атомов, одной из первых его задач было выяснение судьбы запасных жиров в организме животного. Считалось, что животные для удовлетворения своих энергетических потребностей окисляют свежепоглощенные жиры. Жировые отложения рассматривались как резерв, используемый лишь по мере обеднения диеты. Шонхеймер скармливал мышам, поддерживая при этом их постоянный вес, льняное масло, двойные связи жирных кислот которого были насыщены изотопом водорода — дейтерием. Эксперимент показал, что меченые жирные кислоты откладывались в запасных жирах и что последние постоянно участвуют в обмене веществ. В целом благодаря исследованиям Шонхеймера с мечеными углеводородами и аминокислотами было установлено, что все компоненты тела находятся в весьма активном динамическом состоянии, образуя так называемый «метаболический котел».
Если при первых опытах Шонхеймера принципы работы этого котла были еще не ясны, то обнаружение тесной связи между обменом липидов и аминокислот, с одной стороны, и углеводов (циклом трикарбоновых кислот), с другой, позволили впервые — сначала лишь гипотетически — наметить контуры сложной динамической системы обратимых и необратимых процессов взаимопревращения промежуточных продуктов обмена веществ и использования энергии в организме.
Таким образом, исследования обмена веществ и его энергетики, т. е. динамическое направление в биологической химии, к 40-м годам XX в. стало главной линией развития этой отрасли биологии. В наши дни открытие новых составных биохимических компонентов организмов и уточнение строения и свойств ранее открытых продолжается.
Изучение биологически активных соединений — ферментов и антибиотиков. Создание новых методов.
В 20-40-х годах была установлена структура, исследована биологическая активность и проведены синтезы важнейших гормонов (Л. Ружичка и А. Бутенандт, Нобелевская премия, 1939), стероидов и желчных кислот (А. Виндаус, Нобелевская премия, 1928; Г. Виланд, Нобелевская премия, 1927), витаминов и коферментов (А. Виндаус, П. Каррер, Р. Кун, П. Стинбок, А. Сцент-Дьёрдьи). Конец 40-х годов ознаменовался открытием антибиотиков — пенициллина (А. Флеминг, Г. Флори и Э. Чейн, Нобелевская премия, 1945) и стрептомицина (3. Ваксман, Нобелевская премия, 1952) и созданием антибиотической промышленности (см. главу 7).
Наиболее важными и тесно связанными с изучением обмена веществ были исследования ферментов как индивидуальных химических соединений. Существовавшие до 20-х годов XX в. сомнения относительно их белковой природы были рассеяны после разработки американским биохимиком Дж. Самнером (1926) метода кристаллизации ферментов и получения кристаллической уреазы. В следующем десятилетии было получено большое число кристаллических ферментов (Дж. Самнер, Дж. Нортроп и У. Стенли — Нобелевская премия, 1946) и все они оказались белками. Новый метод получения чистых препаратов ферментов значительно ускорил исследования кинетики ферментативных реакций и свойств отдельных ферментов.
В 40-е годы начинают интенсивно разрабатываться новые методы исследований, в первую очередь физико-химические. Они знаменовали начало радикальных изменений в осуществлении исследовательского поиска и создание новой экспериментальной техники. Возможность использования автоматического и полуавтоматического лабораторного оборудования, созданного в 50-х годах с использованием принципов, разработанных в предвоенные годы, вызвала бурный рост биологической химии, который она переживает в настоящее время.
В 20-х годах Т. Сведбергом (Нобелевская премия, 1926) были построены первые аналитические ультрацентрифуги с масляными роторами, представлявшие собой гигантские сооружения. С их помощью были получены первые сведения о форме и размерах белковых молекул. В 30-е годы А. Тизелиус (Нобелевская премия, 1948) разработал метод электрофореза в свободной фазе, блестяще примененный Г. Теореллем (Нобелевская премия, 1955) для исследования природы взаимодействия кофермента и апофермента. В 1940 г. А. Мартин и Р. Синг (Нобелевская премия, 1952), использовав идею, высказанную в 1913 г. русским физиологом М.С. Цветом, разработали метод хроматографии на бумаге. Этот принцип, введенный впоследствии и в электрофоретические исследования, произвел настоящую революцию в аналитической биохимии.
Биоэнергетика.
Понятие «биоэнергетика» ввел в науку А. Сцент-Дьёрдьи, выпустивший в 1957 г. монографию под тем же названием. Термин «биоэнергетика» признан наилучшим 11 лет спустя группой ведущих специалистов по биохимии дыхания и фотосинтеза, собравшихся в Полиньяно (Италия), чтобы определить новую область биологии, связанную с изучением молекулярных механизмов энергетического обмена клетки.
Проникновение на молекулярный уровень организации обличает биоэнергетиков от физиологов и биохимиков, изучавших внешние суммарные показатели и спецификацию химических реакций энергетического обмена организма. Биоэнергетика ставит перед собой два вопроса: какие именно молекулы среди всего разнообразия природных соединений ответственны за превращение энергии в клетке, и каким образом они выполняют свою функцию.
Первая из упомянутых проблем уже решена применительно к большинству биологических процессов трансформации энергии. Начало этому направлению было положено в середине 40-х годов, когда В.А. Энгельгардт и М.Н. Любимова обнаружили АТФазную активность нерастворимого мышечного белка миозина. Авторы сделали вывод о том, что именно миозин ответствен за трансформацию химической энергии АТФ в механическую работу мышц. Впоследствии эта мысль получила полное подтверждение в работах многих исследователей, показавших, что гидролиз АТФ вызывает конформационное изменение актомиозинового комплекса, что в свою очередь вызывает сокращение мышечного волокна. В дальнейшем выяснилось, что и многие другие типы трансформации энергии в клетке осуществляются посредством нерастворимых белковых комплексов, которые обычно встроены в те или иные мембранные образования.
Последнее обстоятельство определяет тесную взаимосвязь и взаимопроникновение биоэнергетики и биологии мембран как новой отрасли современной биологии.
В 1949 г. А. Ленинджер в США показал, что важнейший процесс аккумуляции энергии — окислительное фосфорилирование, открытое в 1930 г. В.А. Энгельгардтом, — локализован в митохондриях, имеющих две мембраны — внешнюю и внутреннюю. В начале 60-х годов выяснилось, что внутренняя мембрана митохондрий служит носителем ферментов окислительного фосфорилирования. Вслед за этим было показано, что фотофосфорилирование, обнаруженное в середине 50-х годов Д. Арноном (США), также локализовано в структурных мембранах, а именно в тилакоидах хлоропластов. Мембранная локализация ферментов окислительного фосфорилирования и фотофосфорилирования оказалась характерной для микроорганизмов.
Роль мембран в окислительном фосфорилировании представлялась неясной в рамках традиционных «химических» гипотез энергетического сопряжения, постулировавших, что процессы переноса электронов и образования АТФ связаны между собой через промежуточный продукт неизвестной химической природы. Подобные взгляды развивались в 40-е годы Ф. Липманом (Нобелевская премия, 1953), а затем Э. Слейтером и Б. Чансом (США) и др.
Дальнейший прогресс в разработке проблемы окислительного фосфорилирования связан с именем английского биохимика П. Митчела, разработавшего в 1961–1966 гг. так называемую хемиосмотическую теорию окислительного фосфорилирования. По Митчелу, химическая энергия процесса окисления в митохондриях превращается сначала в электрическую (мембранный потенциал), а затем вновь переходит в химическую форму и используется для фосфорилирования АТФ неорганическим фосфатом. В самые последние годы эта гипотеза была подтверждена работами советских и американских лабораторий. Оказалось, что цепь окислительных ферментов — переносчиков электронов располагается поперек внутренней мембраны митохондрий, в результате чего окислительная реакция приводит к переносу электронов от одной стороны мембраны к другой ее стороне и к появлению разности электрических потенциалов между вне- и внутримитохондриальными участками. Мембранный потенциал может быть образован также за счет гидролиза АТФ митохондриальной АТФазой, локализованной во внутренней мембране. Этот процесс обратим, что обеспечивает возможность использования энергии окисления, превращенной в мембранный потенциал, для синтеза АТФ.
В 1971 г. Э. Ракеру (США) удалось продемонстрировать самосборку мембранных пузырьков, способных к окислительному фосфорилированию. Пузырьки, образованные из фосфолипидов и очищенных ферментов дыхания и фосфорилирования, генерировали мембранный потенциал и синтезировали АТФ за счет энергии, освобождающейся при окислении аскорбиновой кислоты кислородом.
Изучение функций мембранного потенциала в митохондриях привело к выяснению природы осмотической работы целого ряда биомембран. Оказалось, что катионы, проникающие через мембрану митохондрий, накапливаются внутри этих органелл под действием электрического поля, которое генерируется дыханием или расщеплением АТФ. Обнаружилось также, что проникающие слабые кислоты аккумулируются митохондриями, двигаясь по градиенту pH, возникающему в результате работы тех же ферментативных систем, которые образуют мембранный потенциал.
Выяснение природы движущих сил ионного транспорта в митохондриях позволило продвинуться в понимании механизма осмотической работы мембраны хлоропластов и бактерий. Оказалось, что и в этих случаях химическая энергия субстратов окисления или АТФ, а также энергия света сначала превращается в электрическую и лишь затем используется для переноса целого ряда соединений против концентрационных градиентов.
Иной механизм обнаружился при изучении осмотической работы клеточной мембраны животных организмов. Оказалось, что в этом случае первичным процессом является, как правило, выход из клетки ионов Na+ в обмен на внешние ионы K+. Источником энергии служит гидролиз АТФ, который катализируется специальным мембранным ферментом — АТФазой, активизируемой ионами Na+ и К+ (Na, К-АТФазой). Перенос веществ в клетку против концентрационных градиентов происходит вместе с ионами Na+, которые движутся внутрь клетки по градиенту своей концентрации. Механизм этого явления, как и строение Na, К-АТФазы, остаются неясными.
Подобное положение характерно также для многих других явлений трансформации энергии в клетке, таких, как механическая работа актомиозина, сократительных белков сперматозоидов, бактерий и фагов, механическая работа рибосом, энергообеспечение процесса нервного возбуждения и т. д. Во всех этих случаях выяснена природа ферментов (или комплексов ферментов с небелковыми компонентами), осуществляющих акт трансформации энергии, но каким образом происходит этот акт, пока остается неизвестным. Сказанное относится даже к такому сравнительно простому случаю, как рассеяние энергии дыхания в виде тепла. Доказано, что этот процесс у теплокровных животных может оказываться биологически полезным в условиях внешнего охлаждения организма. Однако в цепи событий, происходящих между дыханием и повышенной продукцией тепла на холоду, еще остаются неясные моменты. Выяснение механизма действия ферментных систем — трансформаторов энергии является важнейшей задачей биоэнергетики на данном этапе ее развития.
* * *
Расцвет биологической химии после первой мировой войны выдвинул ее в число наиболее быстро развивающихся и перспективных, областей знания.
К концу 50-х годов биологическая химия приобрела черты сложной комплексной науки, выводы которой имеют первостепенное значение для обширного круга вопросов. Для изучения закономерностей биохимических процессов и проникновения в сущность жизненных явлений биохимики используют достижения физики, общей, органической и физической химии, фармакологии, патофизиологии и других отраслей медицины, развитие которых теперь в свою очередь зависит от успехов биологической химии.
В рамках этой дисциплины началась дифференциация. Уточнение данных о деталях превращений химических веществ и о различиях в химическом составе организмов привели к возникновению сравнительной и эволюционной биохимии (см. главу 21). Биохимия изучает также важнейшие формы организации материи — пограничную область между живой и неживой природой. В 1924 г. А.И. Опарин выдвинул теорию возникновения жизни на Земле, заложив основы нового направления биохимии (см. главу 22).
Данные биохимии широко Используются в сельском хозяйстве и медицине. Успехи в изучении последовательности процессов основного обмена веществ, прежде всего у микроорганизмов, позволили начиная с 40-х годов приступить к созданию микробиологической промышленности и выпуску антибиотиков, витаминов, органических кислот и аминокислот, некоторых органических полупродуктов и, наконец, кормовых белков (см. главу 7).
Всем этим были заложены основы перехода биологической химии на новый, современный этап, характеризующийся комплексным подходом к изучению процессов, протекающих в организме, в первую очередь био-, синтетических и энергетический, и дальнейшим внедрением в эту науку, методов и представлений физики и физической химии. Развитие исследований по биологической химии, осуществляемых на молекулярном уровне, рассмотрено в главе 23.
Более 800 000 книг и аудиокниг! 📚
Получи 2 месяца Литрес Подписки в подарок и наслаждайся неограниченным чтением
ПОЛУЧИТЬ ПОДАРОК