3.2. Нарушения, влияющие на структуру хроматина в trans-конфигурации
Значение тонко отлаженной структуры хроматина для здоровья человека выдвинулось на первое место в связи с быстро растущим списком заболеваний человека, вызванных мутациями в генах, кодирующих белки, необходимые для структуры и ремоделинга хроматина. Сами по себе эти нарушения не являются эпигенетическими мутациями, но они изменяют состояния хроматина, являющиеся критическими компонентами эпигенотипа. Значительные различия между фенотипами, так же как тот факт, что едва уловимые изменения в уровне белка или даже консервативная аминокислотная замена могут приводить к заболеванию человека, все эти данные уже дают ключ к проблеме, касающейся строго контролируемой регуляции и взаимодействий белков ремоделинга хроматина. Нарушения, которые влияют на хроматин в trans-конфигурации, являются результатом либо нарушения функции белков, непосредственно участвующих в ремоделинге хроматина (таких, как белок, связывающийся с CREB, или СВР, ЕР300, а также белок, связывающийся с метил-CpG, или МеСР2), либо потери функции белков, участвующих в метилировании ДНК (таких, как de novo ДНК-метилтрансфераза ЗВ. DNMT3B, или метилентетрафолатредуктаза, MTHFR) (табл. 23.2). Нарушение функции любого из этих генов вызывает комплексные мультисистемные фенотипы или неоплазии, благодаря эффектам мисрегуляции экспрессии большого числа генов-мишеней, находящихся «ниже по течению». Вполне возможно, что существует множество заболеваний, которые вызываются мутациями в некодирующих РНК, действующими в trans-конфигурапии, хотя их еще предстоит обнаружить.
Таблица 23.2. Выборочные генетические нарушения, влияющие на структуру хроматина в trans-конфигурации
Синдром Рубинштейна-Тайби
Синдром Рубинштейна-Тайби (RSTS, Rubinstein-Taybi syndrome; OMIM 180849) характеризуется задержкой умственного развития, широкими большими пальцами рук и ног, аномалиями лица, врожденными пороками сердца и повышенным риском образования опухолей. Высокая частота конкордантности у монозиготных близнецов, наряду с несколькими случаями передачи от матери к плоду, предполагают, что это заболевание имеет генетическую основу, и что наиболее вероятно аутосомное доминантное наследование. У некоторых пациентов с RSTS были установлены цитогенетические аномалии, включающие 16р13.3 (Tommerup et al., 1992), которые картируются в участке, содержащем ген белка, связывающегося с CREB (CREBBP, или СВР). Гетерозиготные мутации в CREBBP демонстрируют, что гапло-недостаточность СВР вызывает RSTS (Petrij et al., 1995). СВР впервые был описан как коактиватор реагирующего на с АМР белка CREB При повышении внутриклеточных уровней сАМР протеинкиназа А (РКА) перемещается в ядро и фосфорилирует CREB, что приводит к его активации и связыванию его с элементами ответа на количество сАМР (Мауг and Montminy, 2001). СВР — это крупный белок (~250 кДа) с бромодоменом, который, как показано, связывает РКА-фосфорилированный CREB (Chrivia et al., 1993). СВР, в свою очередь, активирует транскрипцию с промотора, содержащего CRE через ацетилирование всех четырех коровых гистонов в соседних нуклеосомах (Ogryzko et al, 1996). Кроме того, СВР взаимодействует непосредственно с основным транскрипционным фактором TFIIIB через участок на своем карбоксильном конце (Anas et al., 1994; Kwok et al., 1994). Функциональный анализ in vitro одной из миссенс-мутаций СВР (замена Arg-1378 на пролин), вызывающей RSTS, выявил, что эта мутация подавляет гистон-ацетилтрансферазную (HAT) активность СВР (Murata et al., 2001). Эти данные, в сочетании с тем, что мыши, гаплонедостаточные по СВР, имеют ослабленную память и обучаемость, измененную синаптическую пластичность и аномальное ацетилирование хроматина, поддерживают вывод о том, что ослабленная НАТ-активность СВР — это главная причина RSTS фенотипа (Alarcon et al., 2004). В соответствии с ролью, которую играет в заболевании понижение НАТ-активности, находится недавнее открытие того, что мутация во втором гене, рЗОО, кодирующем эффективную HAT и транскрипционный коактиватор, вызывает некоторые случаи RSTS (Roelfsema et al., 2005). Выявление того, что некоторые дефекты синаптической пластичности, как и недостатки обучаемости и памяти у СВР " мышей, могут быть ревертированы с помощью ингибиторов деацетилазы гистонов (HDAC) (Alarcon et al., 2004), вызывает вопрос о том, может ли лекарственная терапия, использующая эти реагенты, устранить некоторые ментальные проблемы при RSTS.
Синдром Рэтта
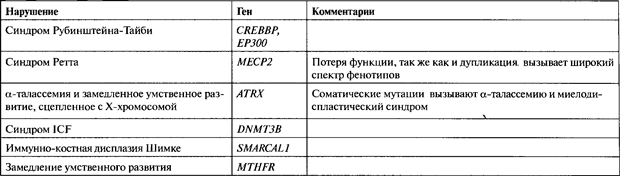
Синдром Рэтта (RTT, Rett syndrome; OMIM 312750) — это доминантное сцепленное с Х-хромосомой постнатальное нарушение развития и нервной системы, характеризующееся двигательными аномалиями, атаксией, припадками, бесцельными движениями рук и речевой регрессией (Hagberg et al., 1983). RTT классифицируется no DSMIV как одно из нарушений, входящих в разряд аутизма (ASD, autistic spectrum disorder), и имеет три основные общие с ASD черты: оба заболевания проявляются постнатально, часто после периода внешне нормального развития, оба нарушают социальное и речевое развитие и оба сопровождаются необычными стереотипными движениями кистей или рук (рис. 23.5а). Хотя RTT — это в подавляющем большинстве случаев (>99%) спорадическое заболевание, обнаружение небольшого числа семей, в которых ген передается по материнской линии, предполагает, что оно имеет генетическую основу. Такие семьи, наряду с тем фактом, что RTT, как правило, наблюдается у особей женского пола, а женщины облигатные носители могут не иметь симптомов, привело к гипотезе, что RTT это доминантное нарушение, сцепленное с Х-хромосомой. С помошью метода «Ап exclusion mapping strategy» ген RTT был локализован в Xq27-qter, и анализ генов-кандидатов (candidate gene analysis) на эту роль указал на ген, кодирующий белок 2, связывающийся с метил-CpG (/МЕСР2), как на ген, вызывающий данное заболевание (Amiretal., 1999).
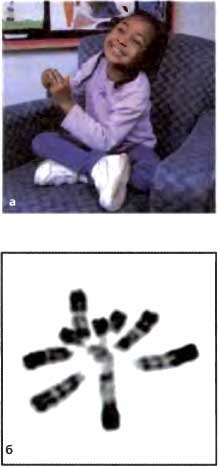
Рис. 5. Генетические нарушения, влияющие на хроматин в cis-конфигурации
(а) Эта фотография пациента с синдромом Рэтта иллюстрирует необычные стереотипные движения рук, жевательные движения и аномальную осанку. Фото любезно предоставлено Д-ром Даниэлем Г . Глэйзом (Dr. Daniel G. Glaze), (б) Микрофотографии хромосом пациента с ICF , любезно предоставленные Д-рами Тимоти Г. Бестором, Робертом А. Роллинсом и Деборой Буркхис (Drs. Timothy Н. Bestor, Robert A. Rollins, and Deborah Bourc'his.)
Обнаружение того, что мутации в МЕСР2 являются основной причиной RTT, служит молекулярным свидетельством наличия связи между RTT и аутизмом. Сейчас известно, что мутации в МЕСР2 вызывают широкий спектр фенотипов у женщин, в том числе неспособность к обучению, обособленное (isolated) замедленное умственное развитие, синдром, похожий на синдром Ангелмана и ASD. Паттерны инактивации Х-хромосомы (XCI) — это главные молекулярные детерминанты данной клинической изменчивости. Женщины с мутациями в МЕСР2 и со сбалансированным паттерном XCI, как правило, страдают классическим RTT, за исключением нескольких гипоморфных аллелей. Женщины с несбалансированными паттернами XCI, благоприятствующими аллели дикого типа, в типичном случае имеют более мягкие фенотипы (Wan et al., 1999; Carney et al., 2003). Мужчины с мутациями MECP2 проявляют более широкий спектр фенотипов, чем женщины, благодаря их гемизиготности по данному локусу. Мутации, вызывающие RTT, как правило, вызывают летальность новорожденных, если только пациент мужского пола не является мозаиком по мутациям или не имеет кариотипа XXY — в этом случае все фенотипы, наблюдаемые у женщин, наблюдаются также и у таких мужчин (Zeev et al., 2002; Neul and Zoghbi, 2004). С другой стороны, у мужчин с гипоморфными аллелями, которые фенотипически едва проявляются у женщин, развиваются любые сочетания признаков, включая умственную отсталость, припадки, треморы, увеличенные семенники, маниакальную депрессию или шизофрению (Meloni et al., 2000; Couvert et al., 2001).
MeCP2 был идентифицирован на основании его способности связываться с симметрично метилированными динуклеотидами CpG (Lewis et al., 1992). Он локализуется в гетерохроматине и действует как репрессор транскрипции, зависимый от метилирования (Nan et al., 1997). МеСР2 связывается с метилированной ДНК через ее метил-CpG-связывающий домен и взаимодействует с корепрессорами Sin3A и HDACs через свой домен транскрипционной репрессии. МеСР2 также ассоциируется с Brahma — компонентом комплекса SWI-SNF ремоделинга хроматина (Harikrishnan et al., 2005).
Интригующей чертой RTT является отсроченное постнатальное проявление фенотипов в отсутствие нейродегенерации. Изучение распределения и распространенности МеСР2 выявило, что он определяется в зрелых нейронах, возможно, после формирования синапсов (Shahbazian et al., 2002а; Kishi and Macklis, 2004; Mullaney et al., 2004). Такое распределение предполагает, что нейронная функция МеСР2 играет существенную роль после того, как установились созревание и активности нейронов, и что эта его функция важна для регуляции деятельности нейронов. Начинается идентификация некоторых мишеней МеСР2, но еще предстоит определить, какими именно из этих мишеней опосредуются разнообразные фенотипы RTT (Chen et al., 2003; Martinowich et al., 2003; Horike et al., 2005; Nuber et al., 2005). Исследования с применением клеточного экстракта от пациентов с RTT или экстракта мозга мышей, не имеющих функционального МеСР2, выявили измененный характер ацетилирования гистонов (Wan et al., 2001; Shahbazian et al., 2002b; Kaufmann et al., 2005), что согласуется с предполагаемой ролью данного белка в деацетилировании гистонов, учитывая его взаимодействия с HDACs. Интересно, что удвоение дозы МеСР2 у мышей и человека приводит к прогрессивным постнатальным фенотипам, которые, в сущности, более резко выражены, чем некоторые из фенотипов, обусловленных потерей функции (Collins et al., 2004; Meins et al., 2005; Van Esch et al., 2005). Остается еще проверить, приводит ли увеличение уровней МеСР2 к титрованию ключевых взаимодействующих компонентов и (или) к аберрантной экспрессии его мишеней Стремясь выявить потенциально новые функции МеСР2, Янг с коллегами обнаружили, что МеСР2 взаимодействует с белком 1, связывающимся с Y-боксом (YB-1), — белком, связывающимся с PF1K, и влияющим на сплайсинг (Young et al., 2005). МеСР2 регулирует сплайсинг РНК репортерных минигенов, но, что наиболее важно, он влияет на сплайсинг РНК in vivo, учитывая данные об изменении паттернов сплайсинга РНК в ткани мозга мышей, моделирующих RTT (Young et al., 2005). Важность МеСР2 в эпигенетической регуляции экспрессии нейрональных генов и его влияние на сплайсинг РНК, по-видимому, лежат в основе потери основных вех при развитии и при аномальной неврологической функции при RTT и родственных ему нарушениях.
Сцепленная с Х-хромосомой ?-талассемия, сопровождающаяся умственной отсталостью
Мужчины с синдромом сцепленной с Х-хромосомой ?-талассемией, сопровождающейся умственной отсталостью (ATRX; OMIM 301040), проявляют ?-талассемию, умеренную (до тяжелой) умственную отсталость, бесформенные черты лица, микроцефалию, аномалии скелета и половых органов и, обычно, неспособность к ходьбе. Гетерозиготные пациенты женского пола обычно не имеют симптомов. Мутации в vmzATRX, которые картируются в Xql3, вызывают этот синдром, так же как и совокупность дополнительных фенотипов, включающих в себя разные степени сцепленной с Х-хромосомой умственной отсталости (XLMR), тяжелую MR со спастической параплегией и благоприобретенную в связи с соматическими мутациями ?-талассемию в миело-диспластическом синдроме (ATMDS) (Gibbons et al., 1995, 2003; Villard et al., 1996; Yntemaet al., 2002). Белок ATRX содержит характерный для растений гомеодоменный (PHD-подобный) мотив «цинкового пальца», равно как и ДНК-зависимую АТФазу семейства SNF2. Это, в сочетании с локализацией в перицентромерных гетерохроматиновых доменах и ассоциацией с гетерохроматином la (HP 1а) (McDowell et al., 1999), предполагает, что данный белок играет какую-то роль в ремоделинге хроматина. Мутации в ATRX вызывают даун-регуляцию локуса ?-глобина и аномальное метилирование нескольких высокоповторяющихся последовательностей нуклеотидов, в том числе субтеломерных повторов, специфичного для Y-хромосомы саттелита и рибосомных последовательностей ДНК. Недавние исследования показали, что ATRX необходим для выживания кортикальных нейронов, и это служит намеком на то, что повышенная потеря нейронов может вносить свой вклад в развитие тяжелой умственной отсталости и мышечной спастичности, наблюдаемых у пациентов с мутациями ATRX (Berube et al., 2005).
Интересно, что уровни ATRX строго регулируются и что как уменьшение, так и увеличение его количества, вызывают основные проблемы в развитии нервной системы. Например, пациенты, у которых в результате мутаций имеется 10—30% ATRX от его нормального уровня, проявляют полный фенотип, характерный для ATRX, несмотря на наличие значительного количества нормального белка ATRX (Picketts et al., 1996). Слишком большое количество ATRX, похоже, тоже является разрушительным. У трансгенных мышей с сверхэкспрессией ATRX развиваются дефекты нервной трубки, наблюдается замедление роста и гибель в процессе эмбриогенеза. У тех, кто выживает, наблюдаются краниофасциальные аномалии, непроизвольное царапанье морды и припадки. Эти характеристики напоминают клиническую картину у пациентов с мутациями ATRX типа «потери функций», что указывает на возможность того, что уровни ATRX строго регулируются для функциональной целостности белкового комплекса, внутри которого он находится.
Синдром иммунодефицита, нестабильности центромерного участка и лицевых аномалий
Синдром иммунодефицита, нестабильности центромерного участка и лицевых аномалий (ICF, OMIM 242860) — это редкое аутосомное рецессивное нарушение, связанное с разрывом хромосом. Пациенты с ICF проявляют два неизменных фенотипа: иммунодефицит и цитогенетические аномалии. Высокоизменчивые и менее пенетрантные фенотипы включают в себя черепно-лицевые дефекты, такие как широкую и плоскую переносицу, складки эпикантуса, высокий лоб и низко посаженные уши, задержку психомоторного развития и кишечную дисфункцию (Smeets et al., 1994). В типичном случае иммунодефицит представлен в тяжелых формах и часто вызывает смерть в детстве, до достижения зрелого возраста, в результате респираторных или желудочно-кишечных инфекций. Наиболее обычный иммунологический дефект — это снижение уровня сывороточных иммуноглобулинов (IgG), но наблюдается также и уменьшение числа В- или Т-клеток (Ehrlich, 2003). Цитогенетические аномалии, в первую очередь затрагивающие хромосомы 1 и 16, и, в меньшей степени, 9, видны при обычном кариотипическом анализе крови и в культивируемых клетках пациентов с ICF (рис. 5b) (Tuck-Muller et al., 2000).
Гипометилирование околоцентромерных повторяющихся последовательностей на хромосомах 1, 9 и 16 было обнаружено задолго до идентификации гена ICF (Jean-pierre et al., 1993). Эти хромосомы содержат наиболее крупные блоки классических сателлитных (сателлиты 2 и 3) тандемных повторов около центромеров. Тот факт, что ICF вызывается мутациями типа «потери функции» в гене ДНК метилтрансферазы (DNMT3B), метилирующей ДНК de novo, способствовало пониманию роли ослабления метилирования в центромерных сателлитах 2 и 3 (Hansen et al., 1999; Okano et al., 1999; Xu et al., 1999). Однако остается неясным, почему потеря функции у повсеместно экспрессирующейся de novo метилтрансферазы избирательно затрагивает специфические повторяющиеся последовательности. Одно из возможных объяснений связано с внутриклеточным распределением и (или) контекст-специфичным белковым взаимодействием DNMT3B (Bachman et al., 2001). Другая возможность состоит в том, что каталитическая активность DNMT3B в большей степени необходима для метилирования последовательностей нуклеотидов с высокой плотностью CpGs на больших геномных участках, как в случае с сателлитом 2 (Gowher and Jeltsch, 2002) или повторяющейся последовательностью D4Z4, что подразумевается при плече-лопаточно-лицевой миопатии (Kondo et al., 2000). Остается определить, являются ли гипометилированными дополнительные специфические последовательности, но прогнозируется, что гипометилирование ДНК ведет к изменению экспрессии генов, которые играют важную роль в развитии черепно-лицевой области, нервной и иммунной систем.
Изучение экспрессии генов с использованием РНК из лимфобластоидных клеточных линий от пациентов с ICF и от здоровых контрольных людей выявило несколько изменений в генах, участвующих в созревании, миграции, активации и хоминге лимфоцитов (Ehrlich et al., 2001). Не понятно, однако, вызывает ли потеря DNMT3B дисрегуляцию таких генов, потому что паттерны метилирования в их промоторах не кажутся измененными. Притом, что единственное выявленное пока гипометилирование при ICF имеет место на саттелитной ДНК, предполагается, что некоторые из генов, измененных при ICF, могут связываться с саттелитной ДНК. Такие нуклеотидные последовательности в типичном случае, будучи метилированными, ведут себя как гетерохроматин; таким образом, при ICF имеет место разрегулированная экспрессия генов, благодаря транс-эффектам гетерохроматиновых участков, обогащенных доменами с сателлитами 2 и 3 (Bickmore and van der Maarel, 2003).
Спондилоэпифизарная дисплазия Шимке
Дисплазия Шимке (SIOD, OMIM 242900) — это аутосомное рецессивное мультисистемное нарушение, характеризующееся дисплазией позвоночника и концов длинных костей, недостаточностью роста, функциональными почечными аномалиями вследствие очагового (центрального) гломерулосклероза, гипотироидизмом и дефектным Т-клеточным иммунитетом (Schimke et al., 1971; Spranger et al., 1991). SIOD вызывается мутациями в SMARCAL1 (SWl/SNF2-CBB3aHHbm, ассоциированный с матриксом; актин-зависимый регулятор хроматина, подсемейство а-like 1), который кодирует белок, регулирующий транскрипционную активность посредством ремоделинга хроматина (Boerkoel et al., 2002). Нонсенс-мутации и мутации сдвига рамки считывания вызывают тяжелые нарушения фенотипа, тогда как некоторые из миссенс-мутаций вызывают возникновение более мягких, или частично выраженных фенотипов (Boerkoel et al., 2002). Недавно у пациента с лимфомой В-клетоки с SIOD была обнаружены мутации в SMARCAL; это предполагает, что потеря функции данного белка может вызывать фатальное лимфопролиферативное расстройство (Taha et al., 2004). Остается прояснить точный механизм, посредством которого утрата SMARCAL1 вызывает фенотип SIOD.
Дефицит метилентетрагидрофолатредуктазы
Метилентетрагидрофолатредуктаза (MTHFR) участвует в превращении 5, 10-метилентетрагидрофолата (5,10-MTHF) в 5-метилтетрафолат (5MTHF). Метальная группа приобретается затем от 5MTHF во время превращения гомоцистеина в метионин с помощью метионинсинтазы. Метионин далее превращается в S-аденозилметионин (SAM), основной донор метила для всех метилтрансфераз. Дефицит MTHFR вызывает редкое аутосомное рецессивное расстройство, характеризующееся умственной отсталостью (Rozen, 1996). Обычный термолабильный полиморфизм (6770Т, который изменяет аланин на валин) вызывает понижение активности MTHFR и ассоциировался, особенно у гомозоготных пациентов, в диете которых низкое содержание фолатов, с гипергомоцистеинемией (Goyette et al., 1994). Этот полиморфизм изучался как фактор риска возникновения атеросклероза, дефектов нервной трубки и рака (Ма et al., 1997; Brattstrometal., 1998; Chenetal., 1999; Bottoand Yang, 2000; Schwahn and Rozen, 2001). Мыши, гетерозиготные или гомозиготные по нулевой аллели MTHFR, имеют пониженный уровень SAM, пониженное общее метилирование ДНК. Более того, такие нуль-мутанты имеют липидные отложения на аорте и дегенерацию нейронов (Chen et al., 2001). Общее изменение метилирования ДНК, связанное с частичной или полной потерей MTHFR, позволяет предположить, что некоторые из фенотипов, ассоциирующиеся с этими дисфункциями, могут являться следствием нарушений хроматина благодаря пониженному метилированию ДНК (и, возможно, гистонов). Имеется одно сообщение о том, что дефицит MTHFR вызывает фенотип, характерный для синдрома Ангелмана (Arn et al., 1998), и имеются существенные совпадения тяжелой недостаточности по MTHFR с AS и RTT (Fattal-Valevski et al., 2000).
Более 800 000 книг и аудиокниг! 📚
Получи 2 месяца Литрес Подписки в подарок и наслаждайся неограниченным чтением
ПОЛУЧИТЬ ПОДАРОК